CLuster comparison
Katharina Hembach
8/26/2020
Last updated: 2020-09-02
Checks: 7 0
Knit directory: neural_scRNAseq/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it's best to always run the code in an empty environment.
The command set.seed(20200522) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version ec1d5a9. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: ._.DS_Store
Ignored: ._Rplots.pdf
Ignored: .__workflowr.yml
Ignored: ._neural_scRNAseq.Rproj
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/._.DS_Store
Ignored: analysis/._01-preprocessing.Rmd
Ignored: analysis/._01-preprocessing.html
Ignored: analysis/._02.1-SampleQC.Rmd
Ignored: analysis/._03-filtering.Rmd
Ignored: analysis/._04-clustering.Rmd
Ignored: analysis/._04-clustering.knit.md
Ignored: analysis/._04.1-cell_cycle.Rmd
Ignored: analysis/._05-annotation.Rmd
Ignored: analysis/._Lam-0-NSC_no_integration.Rmd
Ignored: analysis/._Lam-01-NSC_integration.Rmd
Ignored: analysis/._Lam-02-NSC_annotation.Rmd
Ignored: analysis/._NSC-1-clustering.Rmd
Ignored: analysis/._NSC-2-annotation.Rmd
Ignored: analysis/.__site.yml
Ignored: analysis/._additional_filtering.Rmd
Ignored: analysis/._additional_filtering_clustering.Rmd
Ignored: analysis/._index.Rmd
Ignored: analysis/._organoid-01-clustering.Rmd
Ignored: analysis/._organoid-02-integration.Rmd
Ignored: analysis/._organoid-03-cluster_analysis.Rmd
Ignored: analysis/._organoid-04-group_integration.Rmd
Ignored: analysis/._organoid-05-group_integration_cluster_analysis.Rmd
Ignored: analysis/01-preprocessing_cache/
Ignored: analysis/02-1-SampleQC_cache/
Ignored: analysis/02-quality_control_cache/
Ignored: analysis/02.1-SampleQC_cache/
Ignored: analysis/03-filtering_cache/
Ignored: analysis/04-clustering_cache/
Ignored: analysis/04.1-cell_cycle_cache/
Ignored: analysis/05-annotation_cache/
Ignored: analysis/Lam-01-NSC_integration_cache/
Ignored: analysis/Lam-02-NSC_annotation_cache/
Ignored: analysis/NSC-1-clustering_cache/
Ignored: analysis/NSC-2-annotation_cache/
Ignored: analysis/additional_filtering_cache/
Ignored: analysis/additional_filtering_clustering_cache/
Ignored: analysis/organoid-01-clustering_cache/
Ignored: analysis/organoid-02-integration_cache/
Ignored: analysis/organoid-04-group_integration_cache/
Ignored: analysis/organoid-05-group_integration_cluster_analysis_cache/
Ignored: analysis/sample5_QC_cache/
Ignored: data/.DS_Store
Ignored: data/._.DS_Store
Ignored: data/._.smbdeleteAAA17ed8b4b
Ignored: data/._Lam_figure2_markers.R
Ignored: data/._known_NSC_markers.R
Ignored: data/._known_cell_type_markers.R
Ignored: data/._metadata.csv
Ignored: data/data_sushi/
Ignored: data/filtered_feature_matrices/
Ignored: output/.DS_Store
Ignored: output/._.DS_Store
Ignored: output/._NSC_cluster1_marker_genes.txt
Ignored: output/Lam-01-clustering.rds
Ignored: output/NSC_1_clustering.rds
Ignored: output/NSC_cluster1_marker_genes.txt
Ignored: output/NSC_cluster2_marker_genes.txt
Ignored: output/NSC_cluster3_marker_genes.txt
Ignored: output/NSC_cluster4_marker_genes.txt
Ignored: output/NSC_cluster5_marker_genes.txt
Ignored: output/NSC_cluster6_marker_genes.txt
Ignored: output/NSC_cluster7_marker_genes.txt
Ignored: output/additional_filtering.rds
Ignored: output/figures/
Ignored: output/sce_01_preprocessing.rds
Ignored: output/sce_02_quality_control.rds
Ignored: output/sce_03_filtering.rds
Ignored: output/sce_organoid-01-clustering.rds
Ignored: output/sce_preprocessing.rds
Ignored: output/so_04-group_integration.rds
Ignored: output/so_04_1_cell_cycle.rds
Ignored: output/so_04_clustering.rds
Ignored: output/so_additional_filtering_clustering.rds
Ignored: output/so_integrated_organoid-02-integration.rds
Ignored: output/so_merged_organoid-02-integration.rds
Ignored: output/so_organoid-01-clustering.rds
Ignored: output/so_sample_organoid-01-clustering.rds
Untracked files:
Untracked: Rplots.pdf
Untracked: analysis/Lam-0-NSC_no_integration.Rmd
Untracked: analysis/additional_filtering.Rmd
Untracked: analysis/additional_filtering_clustering.Rmd
Untracked: analysis/organoid-04-group_integration.Rmd
Untracked: analysis/organoid-05-group_integration_cluster_analysis.Rmd
Untracked: analysis/sample5_QC.Rmd
Untracked: data/Homo_sapiens.GRCh38.98.sorted.gtf
Untracked: data/Kanton_et_al/
Untracked: data/Lam_et_al/
Untracked: scripts/
Unstaged changes:
Modified: analysis/Lam-02-NSC_annotation.Rmd
Modified: analysis/_site.yml
Modified: analysis/index.Rmd
Modified: analysis/organoid-02-integration.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/organoid-03-cluster_analysis.Rmd) and HTML (docs/organoid-03-cluster_analysis.html) files. If you've configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | ec1d5a9 | khembach | 2020-09-02 | cluster abundances in organoid integration |
Load packages
library(ComplexHeatmap)
library(cowplot)
library(ggplot2)
library(dplyr)
library(muscat)
library(RColorBrewer)
library(Seurat)
library(SingleCellExperiment)Load data & convert to SCE
so <- readRDS(file.path("output", "so_integrated_organoid-02-integration.rds"))
sce <- as.SingleCellExperiment(so, assay = "RNA")
colData(sce) <- as.data.frame(colData(sce)) %>%
mutate_if(is.character, as.factor) %>%
DataFrame(row.names = colnames(sce))
levels(sce$sample_id) <- c("1NSC", "2NSC", "3NC52", "4NC52", "5NC96", "6NC96",
"H9", "409b2")
## order levels according to experiment timeline (Fig. 1a)
levels(sce$group_id) <- c("P22", "D52", "D96", "iPSCs", "EB", "Neuroectoderm",
"Neuroepithelium", "Organoid-1M", "Organoid-2M",
"Organoid-4M")Cluster-sample counts
# set cluster IDs to resolution 0.4 clustering
so <- SetIdent(so, value = "integrated_snn_res.0.4")
so@meta.data$cluster_id <- Idents(so)
sce$cluster_id <- Idents(so)
(n_cells <- table(sce$cluster_id, sce$sample_id))
1NSC 2NSC 3NC52 4NC52 5NC96 6NC96 H9 409b2
0 1 3 4789 1708 3991 1360 1893 1414
1 4462 4516 630 222 561 266 234 198
2 3 4 890 2415 912 653 1096 1619
3 0 0 539 3319 462 156 208 2537
4 0 0 92 1600 70 5 9 4672
5 15 13 933 1259 661 460 584 493
6 0 0 0 1705 0 0 0 2014
7 1267 1283 14 611 14 8 3 388
8 1032 1004 44 717 32 3 0 552
9 0 0 0 821 0 0 0 2401
10 1 2 40 1360 39 22 23 1663
11 2 5 2 1158 0 0 1 1893
12 722 724 203 322 185 262 246 181
13 728 734 271 96 302 251 184 72
14 25 39 181 870 154 54 87 893
15 0 3 0 1148 2 1 0 821
16 3 4 11 616 11 2 7 979
17 1 2 0 273 0 0 0 408
18 69 72 48 52 42 35 20 28Relative cluster-abundances
fqs <- prop.table(n_cells, margin = 2)
mat <- as.matrix(unclass(fqs))
Heatmap(mat,
col = rev(brewer.pal(11, "RdGy")[-6]),
name = "Frequency",
cluster_rows = FALSE,
cluster_columns = FALSE,
row_names_side = "left",
row_title = "cluster_id",
column_title = "sample_id",
column_title_side = "bottom",
rect_gp = gpar(col = "white"),
cell_fun = function(i, j, x, y, width, height, fill)
grid.text(round(mat[j, i] * 100, 2), x = x, y = y,
gp = gpar(col = "white", fontsize = 10)))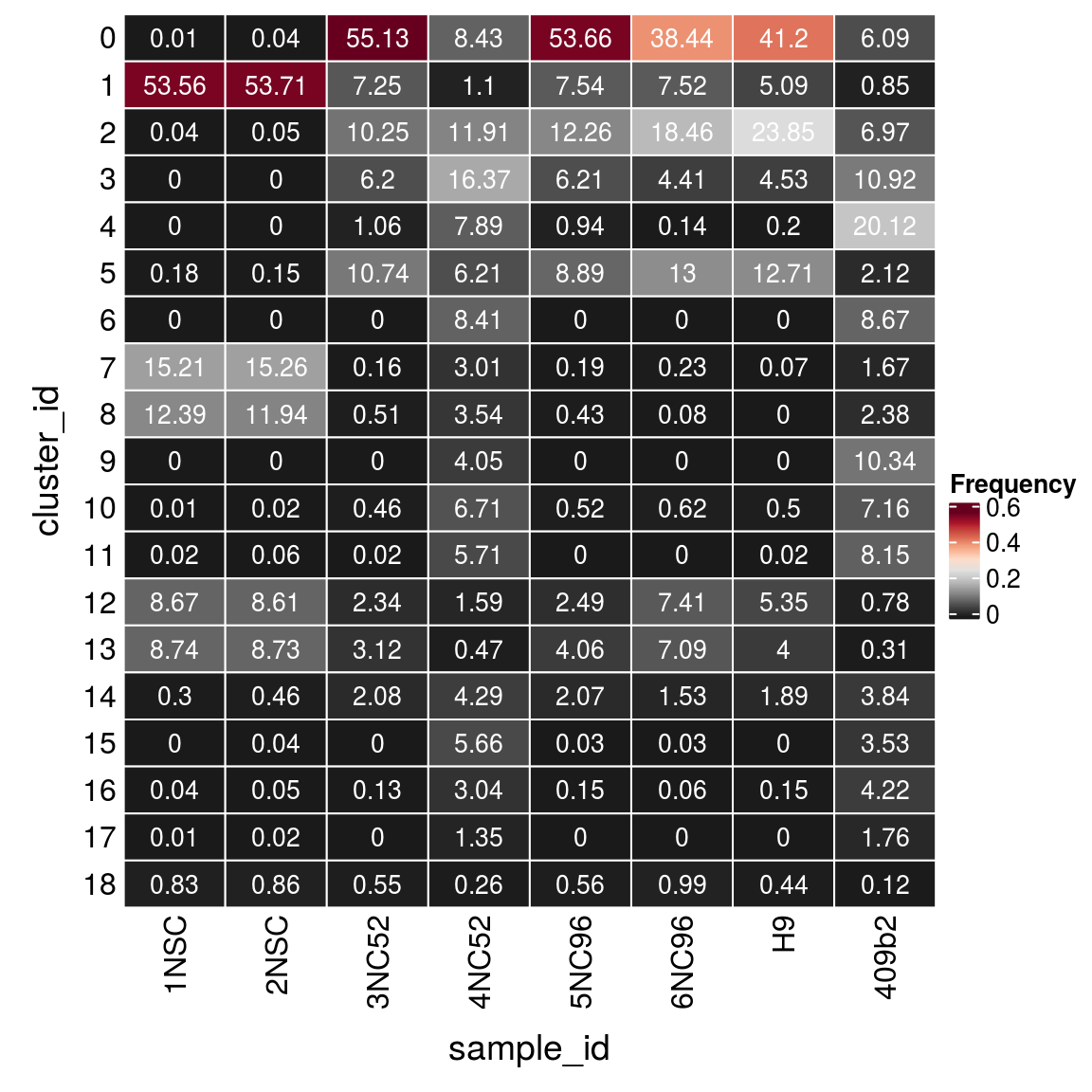
(n_cells_group <- table(sce$cluster_id, sce$group_id))
P22 D52 D96 iPSCs EB Neuroectoderm Neuroepithelium Organoid-1M
0 8780 3253 0 0 0 0 114 2641
1 1191 500 0 0 0 2 52 317
2 1802 1749 0 0 0 0 3 2797
3 1001 364 0 0 0 0 0 3609
4 162 14 0 0 0 1 185 2276
5 1594 1044 0 0 1 0 789 960
6 0 0 30 3651 38 0 0 0
7 28 11 0 2 0 1 26 798
8 76 3 0 0 0 3 371 814
9 0 0 3192 21 9 0 0 0
10 79 45 1 0 87 12 2208 660
11 2 1 18 25 2567 436 5 0
12 388 508 0 0 0 9 156 213
13 573 435 0 0 5 2 61 89
14 335 141 0 0 0 0 9 1151
15 2 1 19 14 13 881 1042 0
16 22 9 0 0 0 0 40 859
17 0 0 40 587 37 16 0 1
18 90 55 0 0 0 0 9 35
Organoid-2M Organoid-4M
0 367 4
1 49 8978
2 1234 7
3 2247 0
4 3810 0
5 2 28
6 0 0
7 172 2550
8 81 2036
9 0 0
10 55 3
11 0 7
12 125 1446
13 11 1462
14 603 64
15 0 3
16 696 7
17 0 3
18 36 141fqs <- prop.table(n_cells_group, margin = 2)
mat <- as.matrix(unclass(fqs))
Heatmap(mat,
col = rev(brewer.pal(11, "RdGy")[-6]),
name = "Frequency",
cluster_rows = FALSE,
cluster_columns = FALSE,
row_names_side = "left",
row_title = "cluster_id",
column_title = "group_id",
column_title_side = "bottom",
rect_gp = gpar(col = "white"),
cell_fun = function(i, j, x, y, width, height, fill)
grid.text(round(mat[j, i] * 100, 2), x = x, y = y,
gp = gpar(col = "white", fontsize = 10)))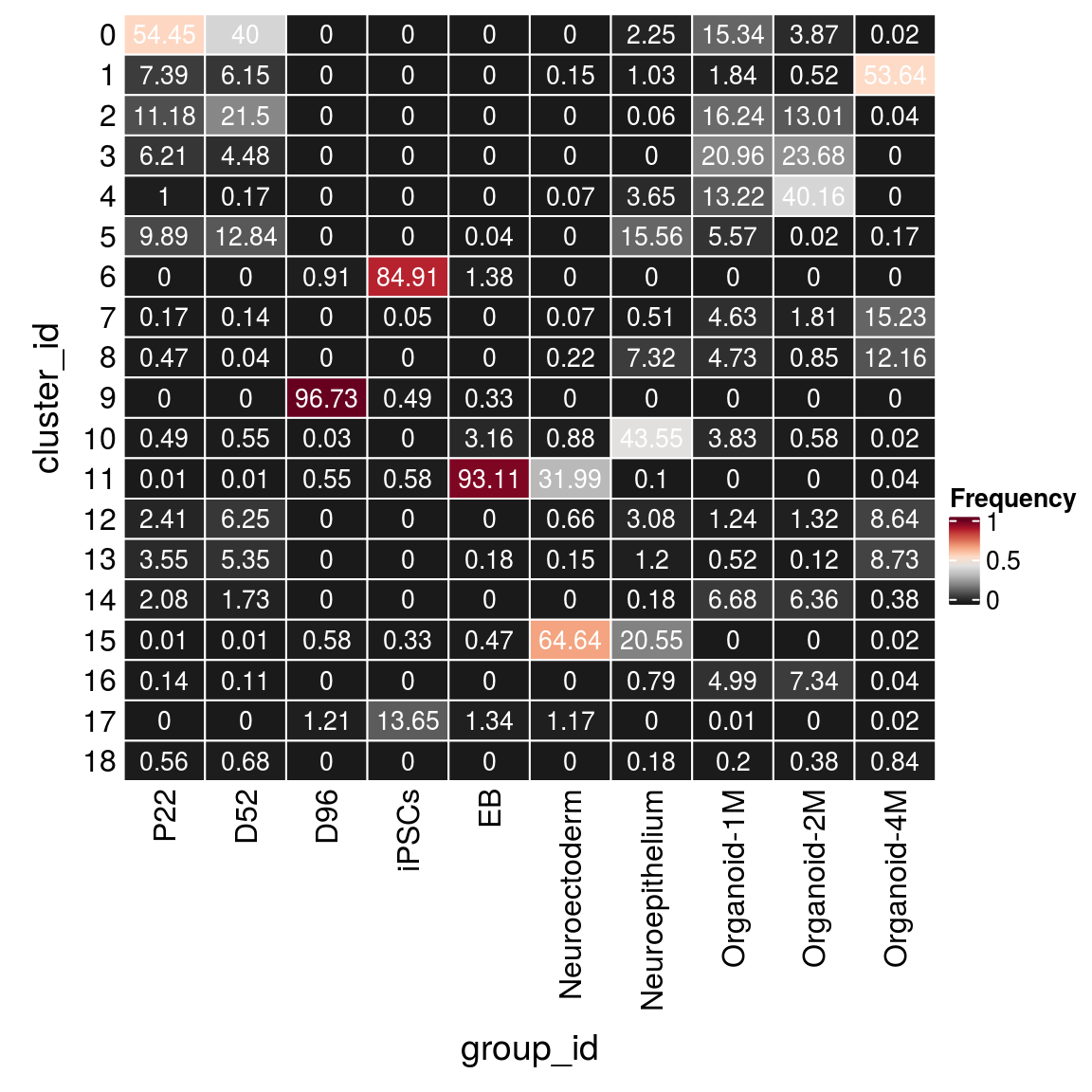
n_cells_lineage <- table(sce$cluster_id, sce$cl_FullLineage)
fqs <- prop.table(n_cells_lineage, margin = 2)
mat <- as.matrix(unclass(fqs))
cn <- colnames(mat)
Heatmap(mat,
col = rev(brewer.pal(11, "RdGy")[-6]),
name = "Frequency",
cluster_rows = FALSE,
cluster_columns = FALSE,
show_column_names = FALSE,
row_names_side = "left",
row_title = "cluster_id",
column_title = "cl_FullLineage",
column_title_side = "bottom",
rect_gp = gpar(col = "white"),
cell_fun = function(i, j, x, y, width, height, fill)
grid.text(round(mat[j, i] * 100, 1), x = x, y = y,
gp = gpar(col = "white", fontsize = 10)),
bottom_annotation = HeatmapAnnotation(
text = anno_text(cn, rot = 80, just = "right")))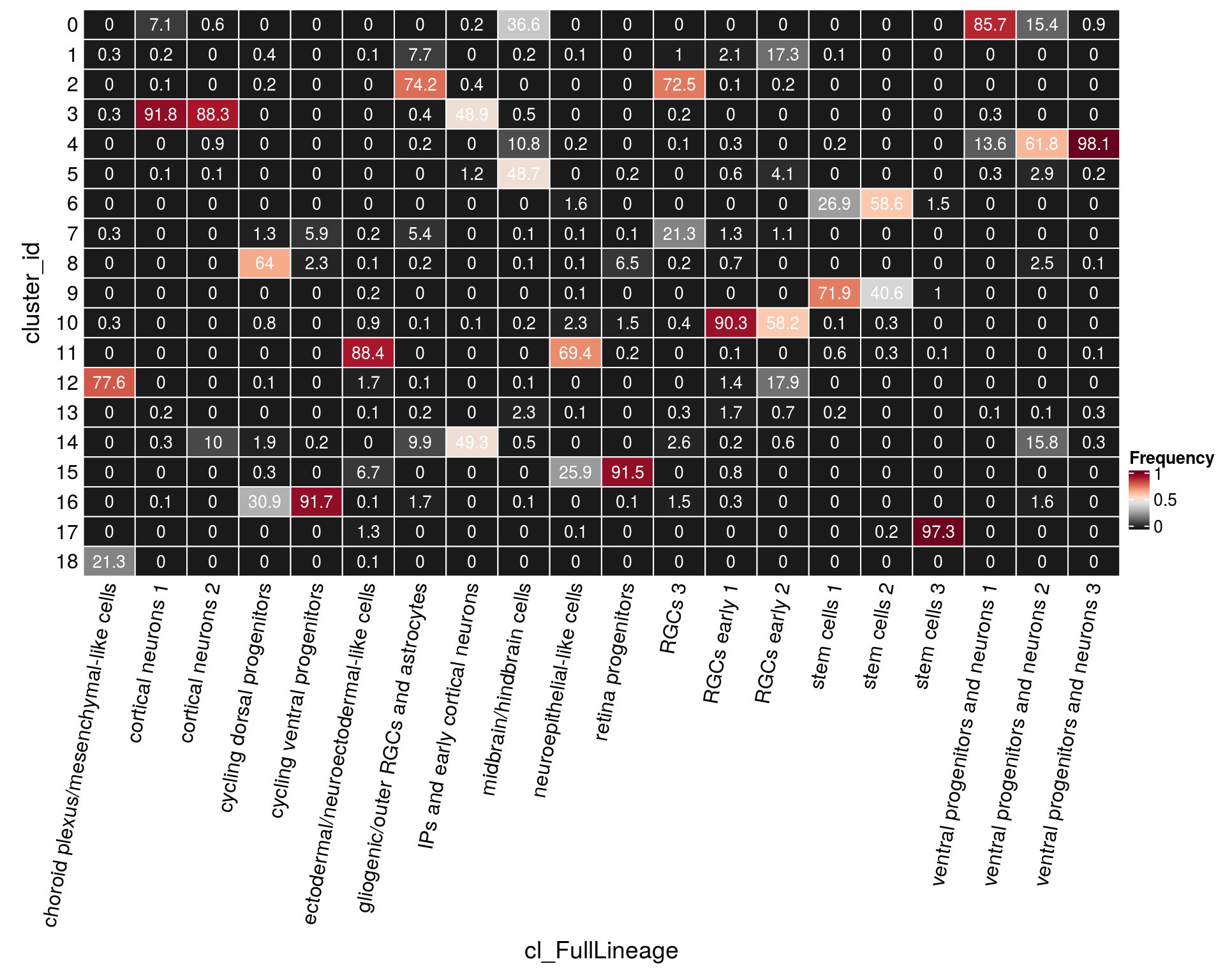
n_cells_lineage <- table(sce$cl_FullLineage, sce$cluster_id)
fqs <- prop.table(n_cells_lineage, margin = 2)
mat <- as.matrix(unclass(fqs))
Heatmap(mat,
col = rev(brewer.pal(11, "RdGy")[-6]),
name = "Frequency",
cluster_rows = FALSE,
cluster_columns = FALSE,
row_names_side = "left",
row_title = "cl_FullLineage",
row_names_rot = 10,
column_title = "cluster_id",
column_title_side = "bottom",
rect_gp = gpar(col = "white"),
cell_fun = function(i, j, x, y, width, height, fill)
grid.text(round(mat[j, i] * 100, 1), x = x, y = y,
gp = gpar(col = "white", fontsize = 10)))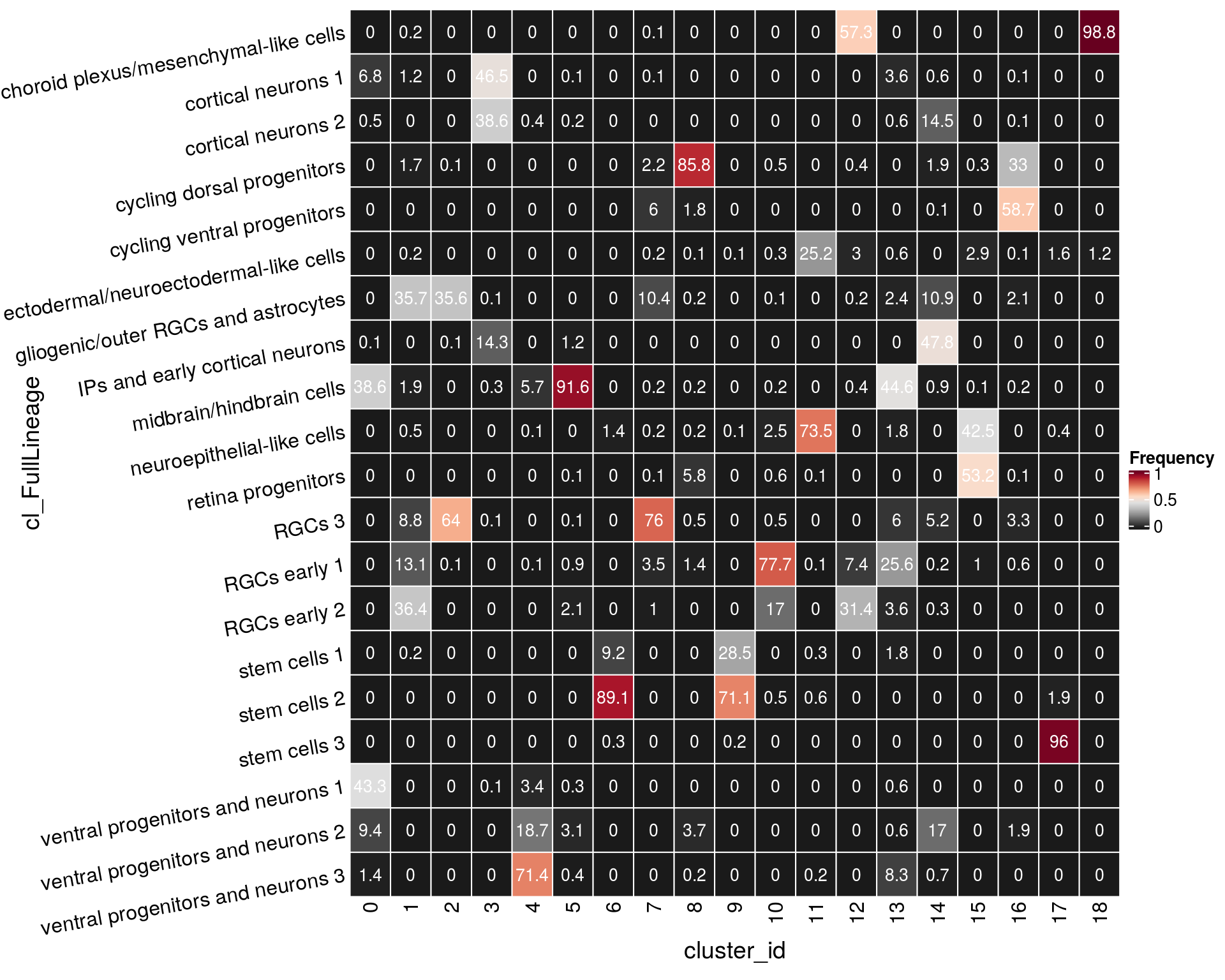
sessionInfo()R version 4.0.0 (2020-04-24)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 16.04.6 LTS
Matrix products: default
BLAS: /usr/local/R/R-4.0.0/lib/libRblas.so
LAPACK: /usr/local/R/R-4.0.0/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 grid stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] SingleCellExperiment_1.10.1 SummarizedExperiment_1.18.1
[3] DelayedArray_0.14.0 matrixStats_0.56.0
[5] Biobase_2.48.0 GenomicRanges_1.40.0
[7] GenomeInfoDb_1.24.2 IRanges_2.22.2
[9] S4Vectors_0.26.1 BiocGenerics_0.34.0
[11] Seurat_3.1.5 RColorBrewer_1.1-2
[13] muscat_1.2.1 dplyr_1.0.0
[15] ggplot2_3.3.2 cowplot_1.0.0
[17] ComplexHeatmap_2.4.2 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] backports_1.1.8 circlize_0.4.10
[3] blme_1.0-4 igraph_1.2.5
[5] plyr_1.8.6 lazyeval_0.2.2
[7] TMB_1.7.16 splines_4.0.0
[9] BiocParallel_1.22.0 listenv_0.8.0
[11] scater_1.16.2 digest_0.6.25
[13] foreach_1.5.0 htmltools_0.5.0
[15] viridis_0.5.1 gdata_2.18.0
[17] lmerTest_3.1-2 magrittr_1.5
[19] memoise_1.1.0 cluster_2.1.0
[21] doParallel_1.0.15 ROCR_1.0-11
[23] limma_3.44.3 globals_0.12.5
[25] annotate_1.66.0 prettyunits_1.1.1
[27] colorspace_1.4-1 rappdirs_0.3.1
[29] ggrepel_0.8.2 blob_1.2.1
[31] xfun_0.15 jsonlite_1.7.0
[33] crayon_1.3.4 RCurl_1.98-1.2
[35] genefilter_1.70.0 lme4_1.1-23
[37] zoo_1.8-8 ape_5.4
[39] survival_3.2-3 iterators_1.0.12
[41] glue_1.4.1 gtable_0.3.0
[43] zlibbioc_1.34.0 XVector_0.28.0
[45] leiden_0.3.3 GetoptLong_1.0.1
[47] BiocSingular_1.4.0 future.apply_1.6.0
[49] shape_1.4.4 scales_1.1.1
[51] DBI_1.1.0 edgeR_3.30.3
[53] Rcpp_1.0.4.6 viridisLite_0.3.0
[55] xtable_1.8-4 progress_1.2.2
[57] clue_0.3-57 reticulate_1.16
[59] bit_1.1-15.2 rsvd_1.0.3
[61] tsne_0.1-3 htmlwidgets_1.5.1
[63] httr_1.4.1 gplots_3.0.4
[65] ellipsis_0.3.1 ica_1.0-2
[67] pkgconfig_2.0.3 XML_3.99-0.4
[69] uwot_0.1.8 locfit_1.5-9.4
[71] tidyselect_1.1.0 rlang_0.4.6
[73] reshape2_1.4.4 later_1.1.0.1
[75] AnnotationDbi_1.50.1 munsell_0.5.0
[77] tools_4.0.0 generics_0.0.2
[79] RSQLite_2.2.0 ggridges_0.5.2
[81] evaluate_0.14 stringr_1.4.0
[83] yaml_2.2.1 knitr_1.29
[85] bit64_0.9-7 fs_1.4.2
[87] fitdistrplus_1.1-1 caTools_1.18.0
[89] RANN_2.6.1 purrr_0.3.4
[91] pbapply_1.4-2 future_1.17.0
[93] nlme_3.1-148 whisker_0.4
[95] pbkrtest_0.4-8.6 compiler_4.0.0
[97] plotly_4.9.2.1 beeswarm_0.2.3
[99] png_0.1-7 variancePartition_1.18.2
[101] tibble_3.0.1 statmod_1.4.34
[103] geneplotter_1.66.0 stringi_1.4.6
[105] lattice_0.20-41 Matrix_1.2-18
[107] nloptr_1.2.2.2 vctrs_0.3.1
[109] pillar_1.4.4 lifecycle_0.2.0
[111] lmtest_0.9-37 GlobalOptions_0.1.2
[113] RcppAnnoy_0.0.16 BiocNeighbors_1.6.0
[115] data.table_1.12.8 bitops_1.0-6
[117] irlba_2.3.3 patchwork_1.0.1
[119] httpuv_1.5.4 colorRamps_2.3
[121] R6_2.4.1 promises_1.1.1
[123] KernSmooth_2.23-17 gridExtra_2.3
[125] vipor_0.4.5 codetools_0.2-16
[127] boot_1.3-25 MASS_7.3-51.6
[129] gtools_3.8.2 DESeq2_1.28.1
[131] rprojroot_1.3-2 rjson_0.2.20
[133] withr_2.2.0 sctransform_0.2.1
[135] GenomeInfoDbData_1.2.3 hms_0.5.3
[137] tidyr_1.1.0 glmmTMB_1.0.2.1
[139] minqa_1.2.4 rmarkdown_2.3
[141] DelayedMatrixStats_1.10.1 Rtsne_0.15
[143] git2r_0.27.1 numDeriv_2016.8-1.1
[145] ggbeeswarm_0.6.0