Cluster annotation
Katharina Hembach
5/29/2020
Last updated: 2020-06-26
Checks: 7 0
Knit directory: neural_scRNAseq/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200522) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version afe57cc. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: ._.DS_Store
Ignored: ._MA.pdf
Ignored: ._MA2.pdf
Ignored: ._MA_plots.pdf
Ignored: ._Rplots.pdf
Ignored: .__workflowr.yml
Ignored: ._hm.pdf
Ignored: ._neural_scRNAseq.Rproj
Ignored: ._sample5_MA_2nd_pop.pdf
Ignored: ._sample5_QC_2nd_pop.pdf
Ignored: ._tmp.pdf
Ignored: ._tmp_detected.pdf
Ignored: ._tmp_manual_discard.pdf
Ignored: ._tmp_manual_discard1.pdf
Ignored: ._tmp_manual_discard_all.pdf
Ignored: ._tmp_manual_discard_all1.pdf
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/._.DS_Store
Ignored: analysis/._01-preprocessing.Rmd
Ignored: analysis/._01-preprocessing.html
Ignored: analysis/._02.1-SampleQC.Rmd
Ignored: analysis/._03-filtering.Rmd
Ignored: analysis/._04-clustering.Rmd
Ignored: analysis/._04-clustering.knit.md
Ignored: analysis/._04.1-cell_cycle.Rmd
Ignored: analysis/._05-annotation.Rmd
Ignored: analysis/.__site.yml
Ignored: analysis/._additional_filtering.Rmd
Ignored: analysis/._additional_filtering_clustering.Rmd
Ignored: analysis/._index.Rmd
Ignored: analysis/01-preprocessing_cache/
Ignored: analysis/02-1-SampleQC_cache/
Ignored: analysis/02-quality_control_cache/
Ignored: analysis/02.1-SampleQC_cache/
Ignored: analysis/03-filtering_cache/
Ignored: analysis/04-clustering_cache/
Ignored: analysis/04.1-cell_cycle_cache/
Ignored: analysis/additional_filtering_cache/
Ignored: analysis/additional_filtering_clustering_cache/
Ignored: analysis/sample5_QC_cache/
Ignored: data/.DS_Store
Ignored: data/._.DS_Store
Ignored: data/._.smbdeleteAAA17ed8b4b
Ignored: data/._metadata.csv
Ignored: data/data_sushi/
Ignored: data/filtered_feature_matrices/
Ignored: output/.DS_Store
Ignored: output/._.DS_Store
Ignored: output/additional_filtering.rds
Ignored: output/figures/
Ignored: output/sce_01_preprocessing.rds
Ignored: output/sce_02_quality_control.rds
Ignored: output/sce_03_filtering.rds
Ignored: output/sce_preprocessing.rds
Ignored: output/so_04_1_cell_cycle.rds
Ignored: output/so_04_clustering.rds
Ignored: output/so_additional_filtering_clustering.rds
Untracked files:
Untracked: MA.pdf
Untracked: MA2.pdf
Untracked: MA_plots.pdf
Untracked: Rplots.pdf
Untracked: analysis/additional_filtering.Rmd
Untracked: analysis/additional_filtering_clustering.Rmd
Untracked: analysis/sample5_QC.Rmd
Untracked: analysis/tabsets.Rmd
Untracked: hm.pdf
Untracked: sample5_MA_2nd_pop.pdf
Untracked: sample5_QC_2nd_pop.pdf
Untracked: scripts/
Untracked: tmp.pdf
Untracked: tmp_detected.pdf
Untracked: tmp_manual_discard.pdf
Untracked: tmp_manual_discard1.pdf
Untracked: tmp_manual_discard_all.pdf
Untracked: tmp_manual_discard_all1.pdf
Unstaged changes:
Modified: analysis/_site.yml
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/05-annotation.Rmd) and HTML (docs/05-annotation.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 06330b1 | khembach | 2020-06-22 | Build site. |
| Rmd | f349423 | khembach | 2020-06-21 | regress out number of UMIs and perc mitochondrial features; cyclone |
| html | 4e52d16 | khembach | 2020-06-15 | Build site. |
| Rmd | 47f9578 | khembach | 2020-06-15 | fix tabset |
| html | d04635a | khembach | 2020-06-12 | Build site. |
| Rmd | f376d76 | khembach | 2020-06-12 | add barplot with sample fraction to heatmap |
| html | 34b86b8 | khembach | 2020-06-12 | Build site. |
| Rmd | 38c33d8 | khembach | 2020-06-12 | do not print marker ids |
| html | 98d3f0d | khembach | 2020-06-12 | Build site. |
| Rmd | a8f31cd | khembach | 2020-06-12 | analyze known marker genes |
| html | f116d0f | khembach | 2020-06-10 | Build site. |
| Rmd | b6767a6 | khembach | 2020-06-10 | wflow_publish(“analysis/05-annotation.Rmd”, verbose = TRUE) |
| html | 419ac73 | khembach | 2020-06-09 | Build site. |
| html | a4d0e04 | khembach | 2020-05-29 | Build site. |
| Rmd | 97d5a52 | khembach | 2020-05-29 | cluster analysis |
Load packages
library(ComplexHeatmap)
library(cowplot)
library(ggplot2)
library(dplyr)
library(muscat)
library(purrr)
library(RColorBrewer)
library(viridis)
library(scran)
library(Seurat)
library(SingleCellExperiment)
library(stringr)
library(RCurl)
library(BiocParallel)Load data & convert to SCE
so <- readRDS(file.path("output", "so_04_clustering.rds"))
sce <- as.SingleCellExperiment(so, assay = "RNA")
colData(sce) <- as.data.frame(colData(sce)) %>%
mutate_if(is.character, as.factor) %>%
DataFrame(row.names = colnames(sce))Number of clusters by resolution
cluster_cols <- grep("res.[0-9]", colnames(colData(sce)), value = TRUE)
sapply(colData(sce)[cluster_cols], nlevels)integrated_snn_res.0.1 integrated_snn_res.0.2 integrated_snn_res.0.4
9 10 17
integrated_snn_res.0.8 integrated_snn_res.1 integrated_snn_res.1.2
23 28 30
integrated_snn_res.2
35 Cluster-sample counts
# set cluster IDs to resolution 0.4 clustering
so <- SetIdent(so, value = "integrated_snn_res.0.4")
so@meta.data$cluster_id <- Idents(so)
sce$cluster_id <- Idents(so)
(n_cells <- table(sce$cluster_id, sce$sample_id))
1NSC 2NSC 3NC52 4NC52 5NC96 6NC96
0 5184 5397 238 107 152 116
1 0 1 1602 1312 447 576
2 1430 1273 69 55 18 11
3 753 727 301 279 340 363
4 4 5 653 676 500 770
5 1 1 970 870 265 374
6 0 0 979 818 256 414
7 0 0 912 781 270 369
8 159 149 541 578 235 286
9 0 0 793 613 209 255
10 426 474 117 108 94 121
11 243 237 203 261 178 206
12 3 2 400 270 236 265
13 0 1 395 277 127 176
14 12 9 315 251 137 191
15 32 45 150 138 40 83
16 84 87 49 44 34 19Relative cluster-abundances
fqs <- prop.table(n_cells, margin = 2)
mat <- as.matrix(unclass(fqs))
Heatmap(mat,
col = rev(brewer.pal(11, "RdGy")[-6]),
name = "Frequency",
cluster_rows = FALSE,
cluster_columns = FALSE,
row_names_side = "left",
row_title = "cluster_id",
column_title = "sample_id",
column_title_side = "bottom",
rect_gp = gpar(col = "white"),
cell_fun = function(i, j, x, y, width, height, fill)
grid.text(round(mat[j, i] * 100, 2), x = x, y = y,
gp = gpar(col = "white", fontsize = 8)))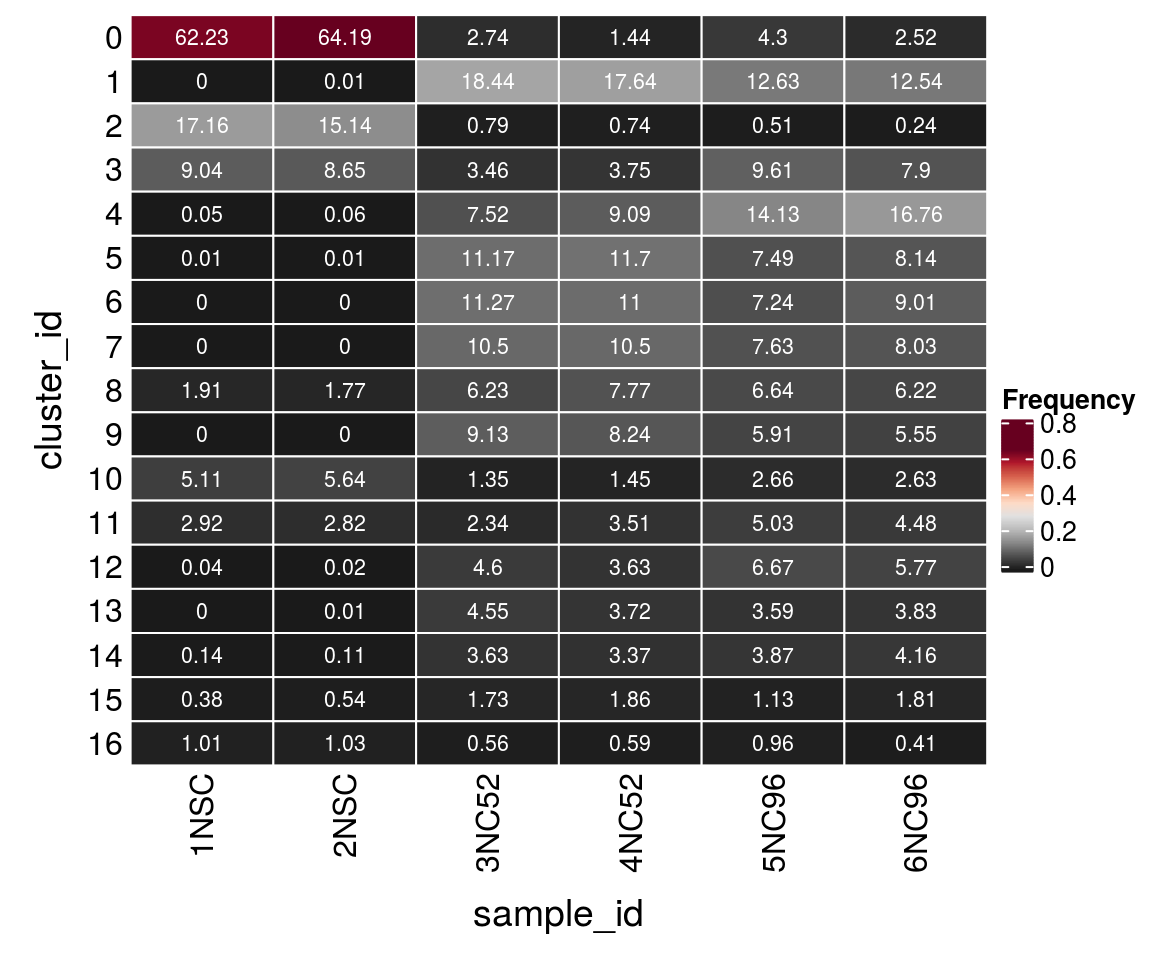
Cell cycle scoring with Seurat
We assign each cell a cell cycle scores and visualize them in the DR plots. We use known G2/M and S phase markers that come with the Seurat package. The markers are anticorrelated and cells that to not express the markers should be in G1 phase.
We compute cell cycle phase:
DefaultAssay(so) <- "RNA"
# A list of cell cycle markers, from Tirosh et al, 2015
cc_file <- getURL("https://raw.githubusercontent.com/hbc/tinyatlas/master/cell_cycle/Homo_sapiens.csv")
cc_genes <- read.csv(text = cc_file)
# match the marker genes to the features
m <- match(cc_genes$geneID[cc_genes$phase == "S"],
str_split(rownames(GetAssayData(so)),
pattern = "\\.", simplify = TRUE)[,1])
s_genes <- rownames(GetAssayData(so))[m]
(s_genes <- s_genes[!is.na(s_genes)]) [1] "ENSG00000012963.UBR7" "ENSG00000049541.RFC2"
[3] "ENSG00000051180.RAD51" "ENSG00000073111.MCM2"
[5] "ENSG00000075131.TIPIN" "ENSG00000076003.MCM6"
[7] "ENSG00000076248.UNG" "ENSG00000077514.POLD3"
[9] "ENSG00000092470.WDR76" "ENSG00000092853.CLSPN"
[11] "ENSG00000093009.CDC45" "ENSG00000094804.CDC6"
[13] "ENSG00000095002.MSH2" "ENSG00000100297.MCM5"
[15] "ENSG00000101868.POLA1" "ENSG00000104738.MCM4"
[17] "ENSG00000111247.RAD51AP1" "ENSG00000112312.GMNN"
[19] "ENSG00000117748.RPA2" "ENSG00000118412.CASP8AP2"
[21] "ENSG00000119969.HELLS" "ENSG00000129173.E2F8"
[23] "ENSG00000131153.GINS2" "ENSG00000132646.PCNA"
[25] "ENSG00000132780.NASP" "ENSG00000136492.BRIP1"
[27] "ENSG00000136982.DSCC1" "ENSG00000143476.DTL"
[29] "ENSG00000144354.CDCA7" "ENSG00000151725.CENPU"
[31] "ENSG00000156802.ATAD2" "ENSG00000159259.CHAF1B"
[33] "ENSG00000162607.USP1" "ENSG00000163950.SLBP"
[35] "ENSG00000167325.RRM1" "ENSG00000168496.FEN1"
[37] "ENSG00000171848.RRM2" "ENSG00000174371.EXO1"
[39] "ENSG00000175305.CCNE2" "ENSG00000176890.TYMS"
[41] "ENSG00000197299.BLM" "ENSG00000198056.PRIM1"
[43] "ENSG00000276043.UHRF1" m <- match(cc_genes$geneID[cc_genes$phase == "G2/M"],
str_split(rownames(GetAssayData(so)),
pattern = "\\.", simplify = TRUE)[,1])
g2m_genes <- rownames(GetAssayData(so))[m]
(g2m_genes <- g2m_genes[!is.na(g2m_genes)]) [1] "ENSG00000010292.NCAPD2" "ENSG00000011426.ANLN"
[3] "ENSG00000013810.TACC3" "ENSG00000072571.HMMR"
[5] "ENSG00000075218.GTSE1" "ENSG00000080986.NDC80"
[7] "ENSG00000087586.AURKA" "ENSG00000088325.TPX2"
[9] "ENSG00000089685.BIRC5" "ENSG00000092140.G2E3"
[11] "ENSG00000094916.CBX5" "ENSG00000100401.RANGAP1"
[13] "ENSG00000102974.CTCF" "ENSG00000111665.CDCA3"
[15] "ENSG00000112742.TTK" "ENSG00000113810.SMC4"
[17] "ENSG00000114346.ECT2" "ENSG00000115163.CENPA"
[19] "ENSG00000117399.CDC20" "ENSG00000117650.NEK2"
[21] "ENSG00000117724.CENPF" "ENSG00000120802.TMPO"
[23] "ENSG00000123485.HJURP" "ENSG00000123975.CKS2"
[25] "ENSG00000126787.DLGAP5" "ENSG00000129195.PIMREG"
[27] "ENSG00000131747.TOP2A" "ENSG00000134222.PSRC1"
[29] "ENSG00000134690.CDCA8" "ENSG00000136108.CKAP2"
[31] "ENSG00000137804.NUSAP1" "ENSG00000137807.KIF23"
[33] "ENSG00000138160.KIF11" "ENSG00000138182.KIF20B"
[35] "ENSG00000138778.CENPE" "ENSG00000139354.GAS2L3"
[37] "ENSG00000142945.KIF2C" "ENSG00000143228.NUF2"
[39] "ENSG00000143401.ANP32E" "ENSG00000143815.LBR"
[41] "ENSG00000148773.MKI67" "ENSG00000157456.CCNB2"
[43] "ENSG00000158402.CDC25C" "ENSG00000164104.HMGB2"
[45] "ENSG00000169607.CKAP2L" "ENSG00000169679.BUB1"
[47] "ENSG00000170312.CDK1" "ENSG00000173207.CKS1B"
[49] "ENSG00000175063.UBE2C" "ENSG00000175216.CKAP5"
[51] "ENSG00000178999.AURKB" "ENSG00000184661.CDCA2"
[53] "ENSG00000188229.TUBB4B" "ENSG00000189159.JPT1" so <- CellCycleScoring(so, s.features = s_genes, g2m.features = g2m_genes,
set.ident = TRUE)
DefaultAssay(so) <- "integrated"DR colored by cluster ID
cs <- sample(colnames(so), 5e3)
.plot_dr <- function(so, dr, id)
DimPlot(so, cells = cs, group.by = id, reduction = dr, pt.size = 0.4) +
guides(col = guide_legend(nrow = 11,
override.aes = list(size = 3, alpha = 1))) +
theme_void() + theme(aspect.ratio = 1)
ids <- c("cluster_id", "group_id", "sample_id", "Phase")
for (id in ids) {
cat("## ", id, "\n")
p1 <- .plot_dr(so, "tsne", id)
lgd <- get_legend(p1)
p1 <- p1 + theme(legend.position = "none")
p2 <- .plot_dr(so, "umap", id) + theme(legend.position = "none")
ps <- plot_grid(plotlist = list(p1, p2), nrow = 1)
p <- plot_grid(ps, lgd, nrow = 1, rel_widths = c(1, 0.2))
print(p)
cat("\n\n")
}cluster_id
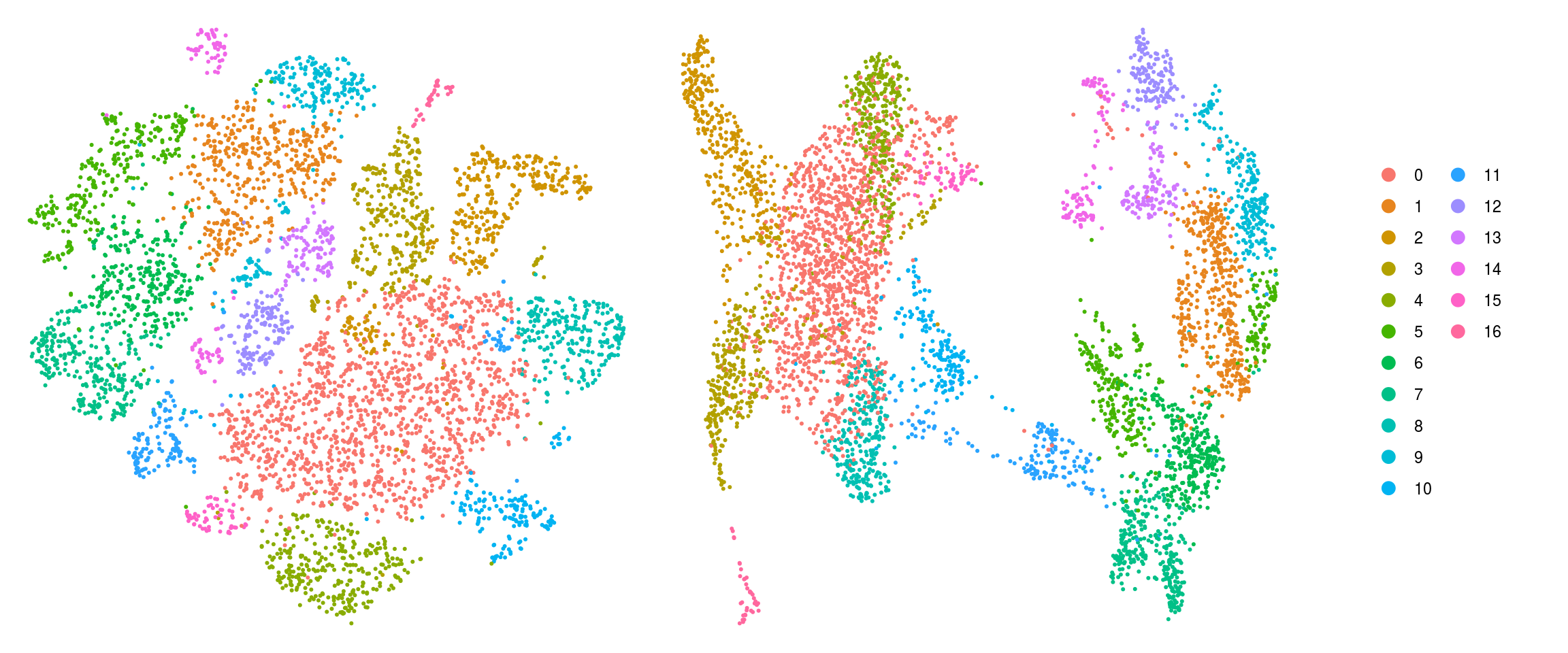
group_id

sample_id


Find markers using scran
We identify candidate marker genes for each cluster that enable a separation of that group from all other groups.
scran_markers <- findMarkers(sce,
groups = sce$cluster_id, block = sce$sample_id,
direction = "up", lfc = 2, full.stats = TRUE)Heatmap of mean marker-exprs. by cluster
We aggregate the cells to pseudobulks and plot the average expression of the condidate marker genes in each of the clusters.
gs <- lapply(scran_markers, function(u) rownames(u)[u$Top == 1])
## candidate cluster markers
lapply(gs, function(x) str_split(x, pattern = "\\.", simplify = TRUE)[,2])$`0`
[1] "SAMD11" "IGFBP5" "PTN" "NEFL" "VIM" "CKB" "TTYH1"
$`1`
[1] "SAMD11" "STMN2" "CRABP1" "ZFHX3" "HOXB8" "MT-CO2"
$`2`
[1] "CENPF" "HMGB2" "VIM" "CKB" "TOP2A"
$`3`
[1] "S100A10" "S100A11" "CLU" "VIM" "CKB" "METRN"
$`4`
[1] "C1orf61" "VCAN" "VIM" "GFAP" "HOXB9" "TTYH1" "MT-ND4"
$`5`
[1] "SAMD11" "STMN2" "HOXB8" "HOXB9" "MT-RNR2"
$`6`
[1] "STMN2" "RTN1" "MEIS2" "HOXB9" "PCP4" "MT-ND3"
$`7`
[1] "FOXP1" "FABP7" "STMN2" "PCP4"
$`8`
[1] "PTPRZ1" "CLU" "LY6H" "VIM" "TAGLN" "DLK1" "METRN"
$`9`
[1] "TAC1" "STMN2" "HOXB5" "MT-CO2"
$`10`
[1] "PTN" "EIF4EBP1" "VIM" "CKB" "FTL"
$`11`
[1] "VGF" "STMN2" "ANXA1" "DDIT3" "HSP90AA1"
$`12`
[1] "POU3F1" "STMN2" "SNCG" "RTN1" "HOXB5" "ONECUT2"
$`13`
[1] "STMN2" "SNCG" "MPPED2" "RTN1" "PCP4"
$`14`
[1] "TAC1" "STMN2"
$`15`
[1] "C1orf61" "HES6" "SOX2" "VIM" "CKB"
$`16`
[1] "S100A11" "COL3A1" "COL1A1" sub <- sce[unique(unlist(gs)), ]
pbs <- aggregateData(sub, assay = "logcounts", by = "cluster_id", fun = "mean")
mat <- t(muscat:::.scale(assay(pbs)))
## remove the Ensembl ID from the gene names
colnames(mat) <- str_split(colnames(mat), pattern = "\\.", simplify = TRUE)[,2]
Heatmap(mat,
name = "scaled avg.\nexpression",
col = viridis(10),
cluster_rows = FALSE,
cluster_columns = FALSE,
row_names_side = "left",
row_title = "cluster_id",
rect_gp = gpar(col = "white"))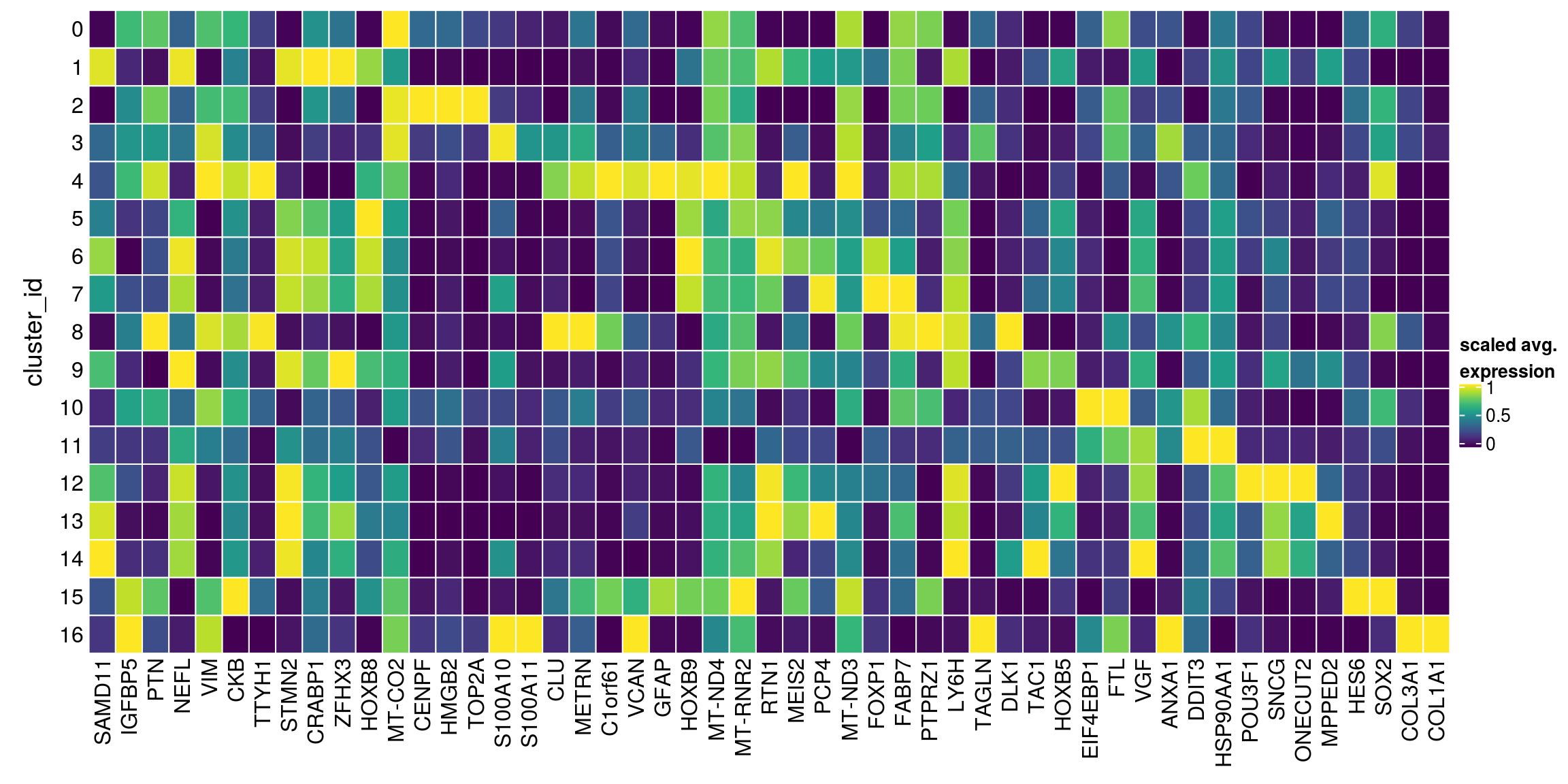
Known marker genes
## source file with list of known marker genes
source(file.path("data", "known_cell_type_markers.R"))
fs <- lapply(fs, sapply, function(g)
grep(pattern = paste0("\\.", g, "$"), rownames(sce), value = TRUE)
)
fs <- lapply(fs, function(x) unlist(x[lengths(x) !=0]) )
gs <- gsub(".*\\.", "", unlist(fs))
ns <- vapply(fs, length, numeric(1))
ks <- rep.int(names(fs), ns)
labs <- lapply(fs, function(x) gsub(".*\\.", "",x))Heatmap of mean marker-exprs. by cluster
# split cells by cluster
cs_by_k <- split(colnames(sce), sce$cluster_id)
# compute cluster-marker means
ms_by_cluster <- lapply(fs, function(gs) vapply(cs_by_k, function(i)
Matrix::rowMeans(logcounts(sce)[gs, i, drop = FALSE]),
numeric(length(gs))))
# prep. for plotting & scale b/w 0 and 1
mat <- do.call("rbind", ms_by_cluster)
mat <- muscat:::.scale(mat)
rownames(mat) <- gs
cols <- muscat:::.cluster_colors[seq_along(fs)]
cols <- setNames(cols, names(fs))
row_anno <- rowAnnotation(
df = data.frame(label = factor(ks, levels = names(fs))),
col = list(label = cols), gp = gpar(col = "white"))
# percentage of cells from each of the samples per cluster
sample_props <- prop.table(n_cells, margin = 1)
col_mat <- as.matrix(unclass(sample_props))
sample_cols <- c("#882255", "#CC6677", "#11588A", "#88CCEE", "#117733", "#44AA99")
sample_cols <- setNames(sample_cols, colnames(col_mat))
col_anno <- HeatmapAnnotation(
perc_sample = anno_barplot(col_mat, gp = gpar(fill = sample_cols),
height = unit(2, "cm"),
border = FALSE),
annotation_label = "fraction of sample\nin cluster",
gap = unit(10, "points"))
col_lgd <- Legend(labels = names(sample_cols),
title = "sample",
legend_gp = gpar(fill = sample_cols))
hm <- Heatmap(mat,
name = "scaled avg.\nexpression",
col = viridis(10),
cluster_rows = FALSE,
cluster_columns = FALSE,
row_names_side = "left",
column_title = "cluster_id",
column_title_side = "bottom",
column_names_side = "bottom",
column_names_rot = 0,
column_names_centered = TRUE,
rect_gp = gpar(col = "white"),
left_annotation = row_anno,
top_annotation = col_anno)
draw(hm, annotation_legend_list = list(col_lgd))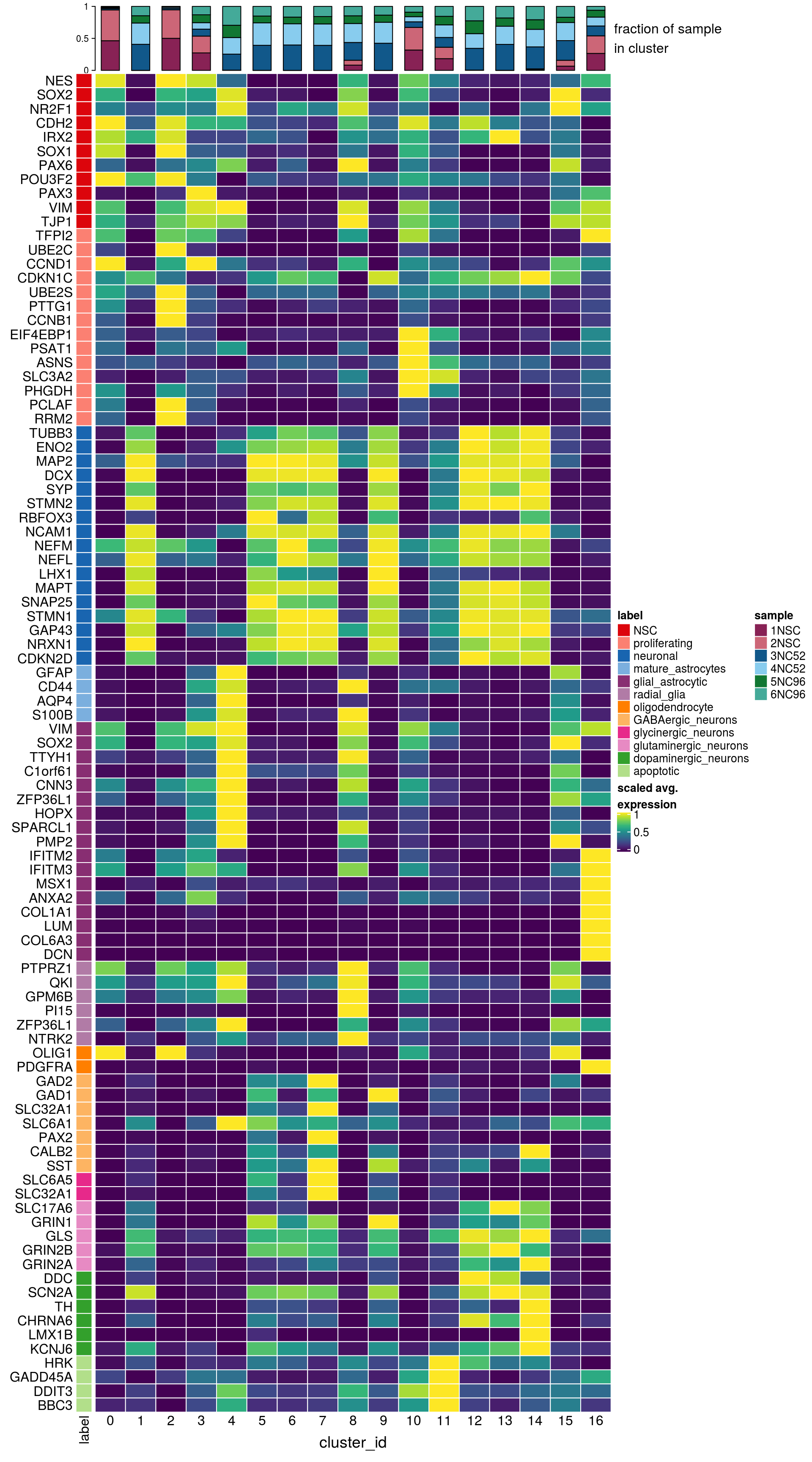
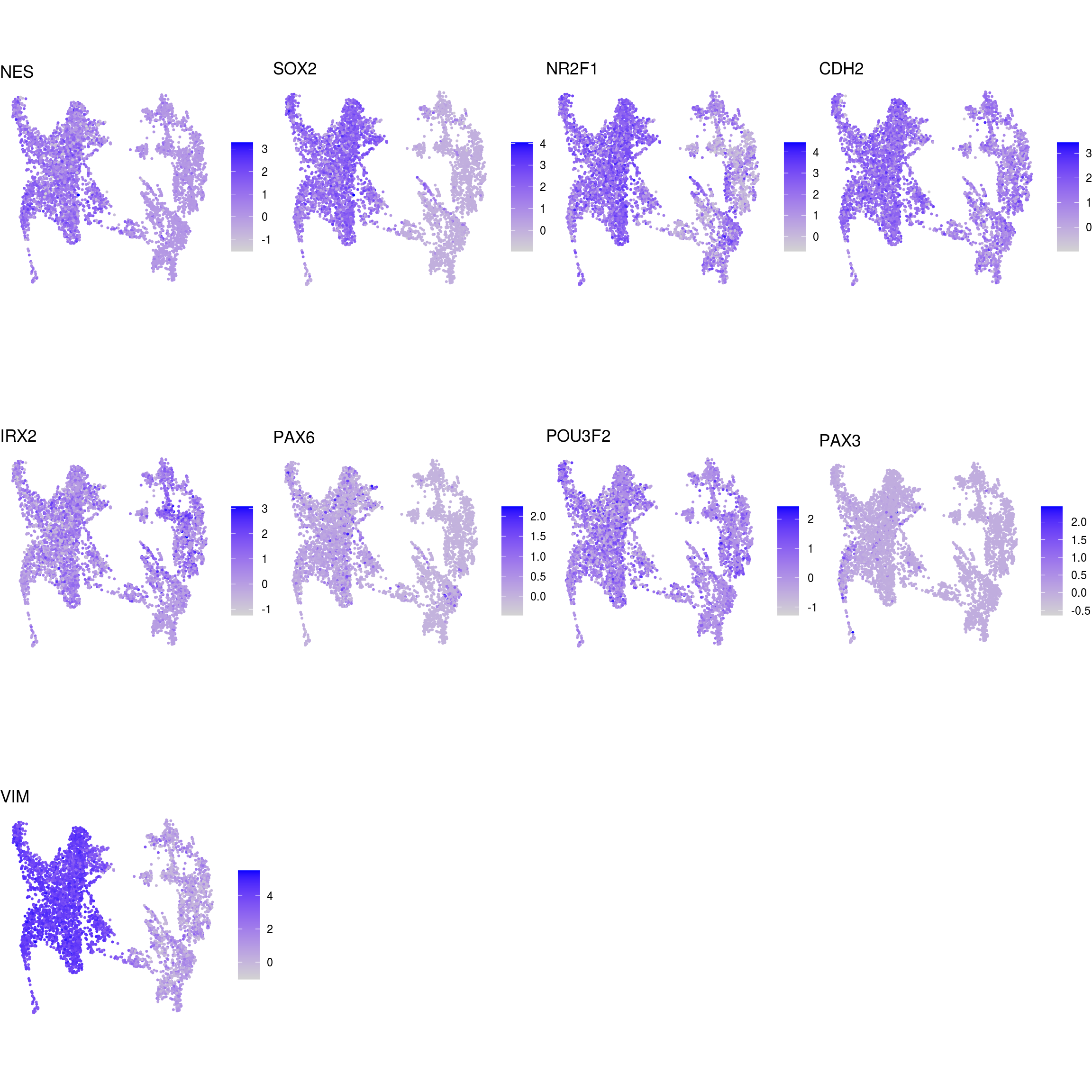
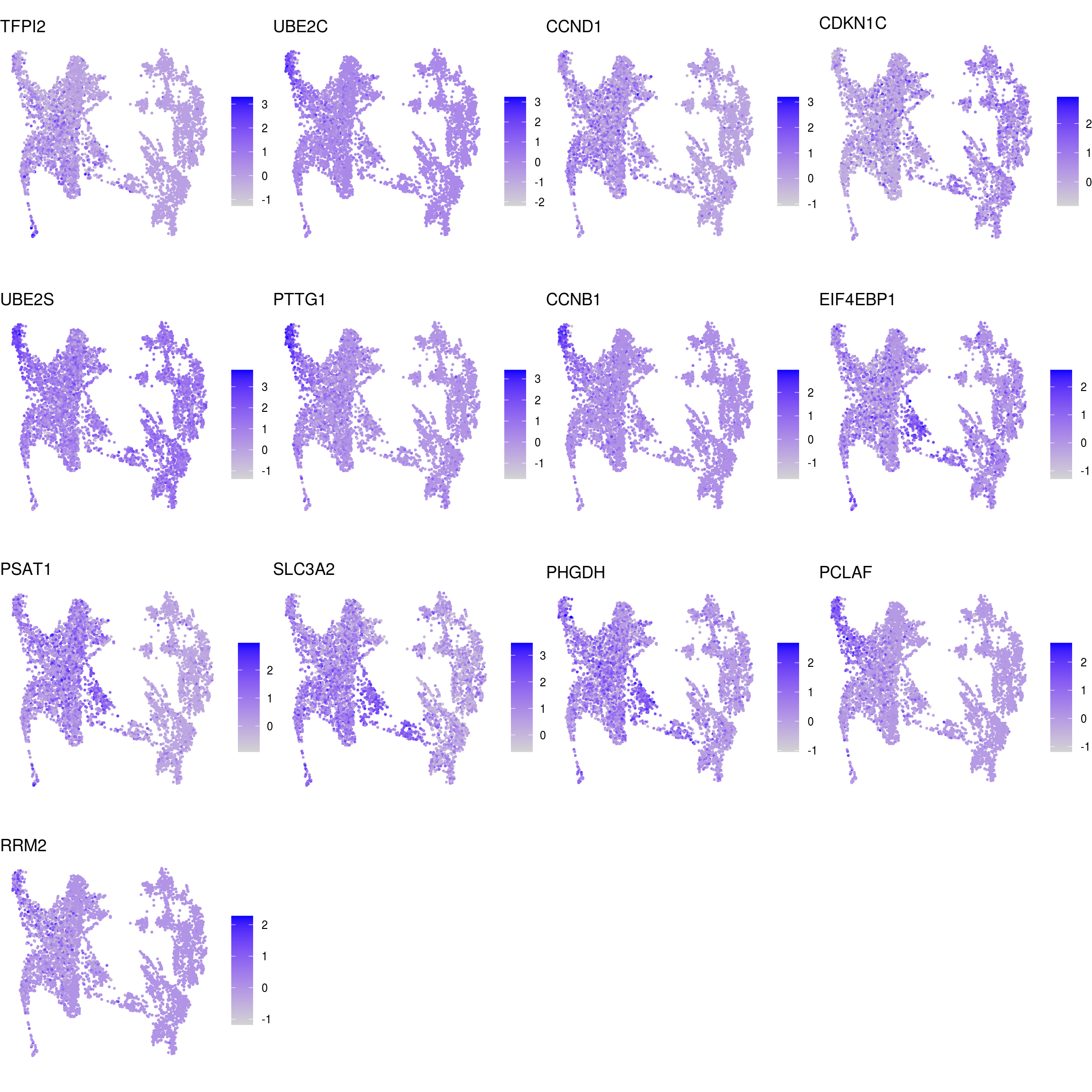
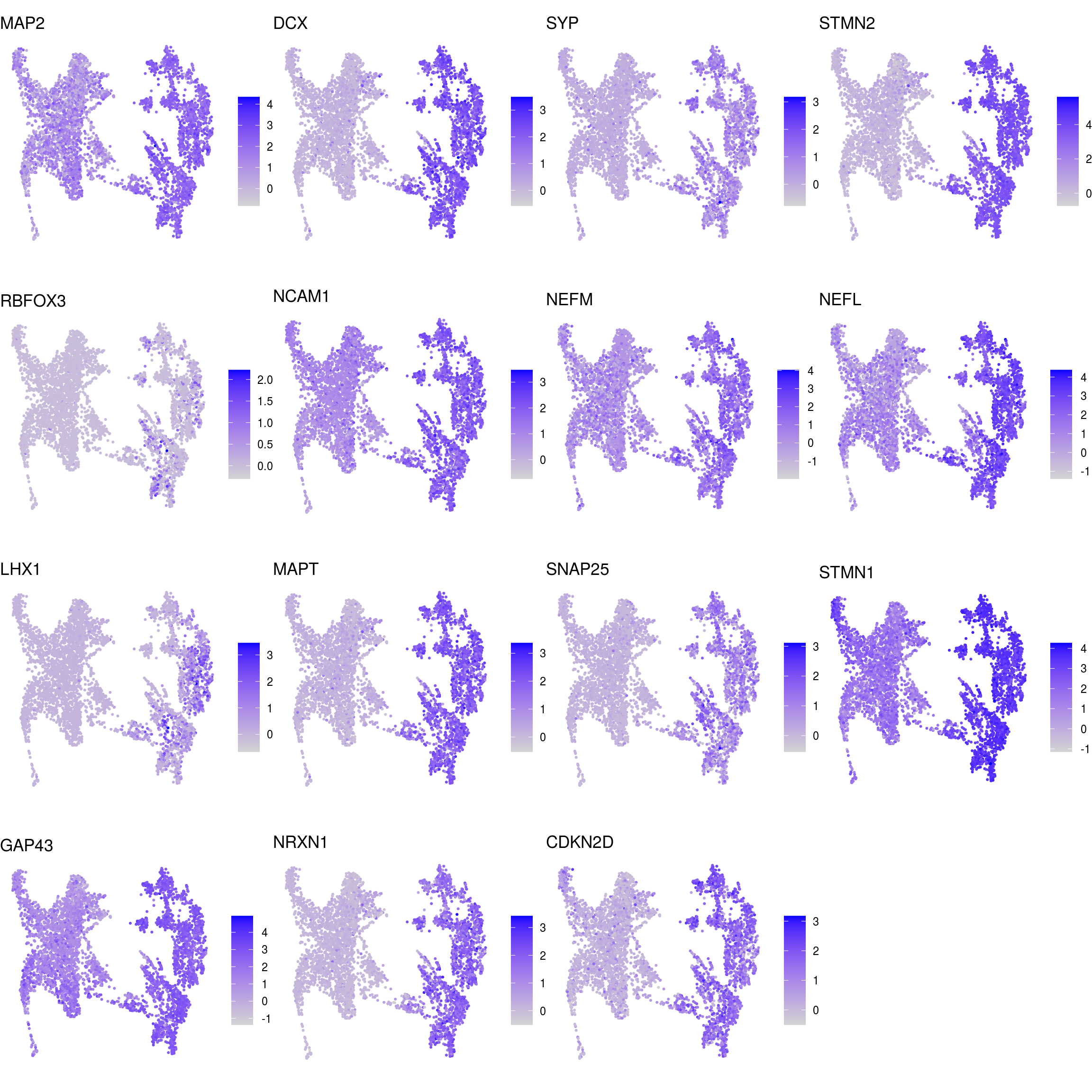
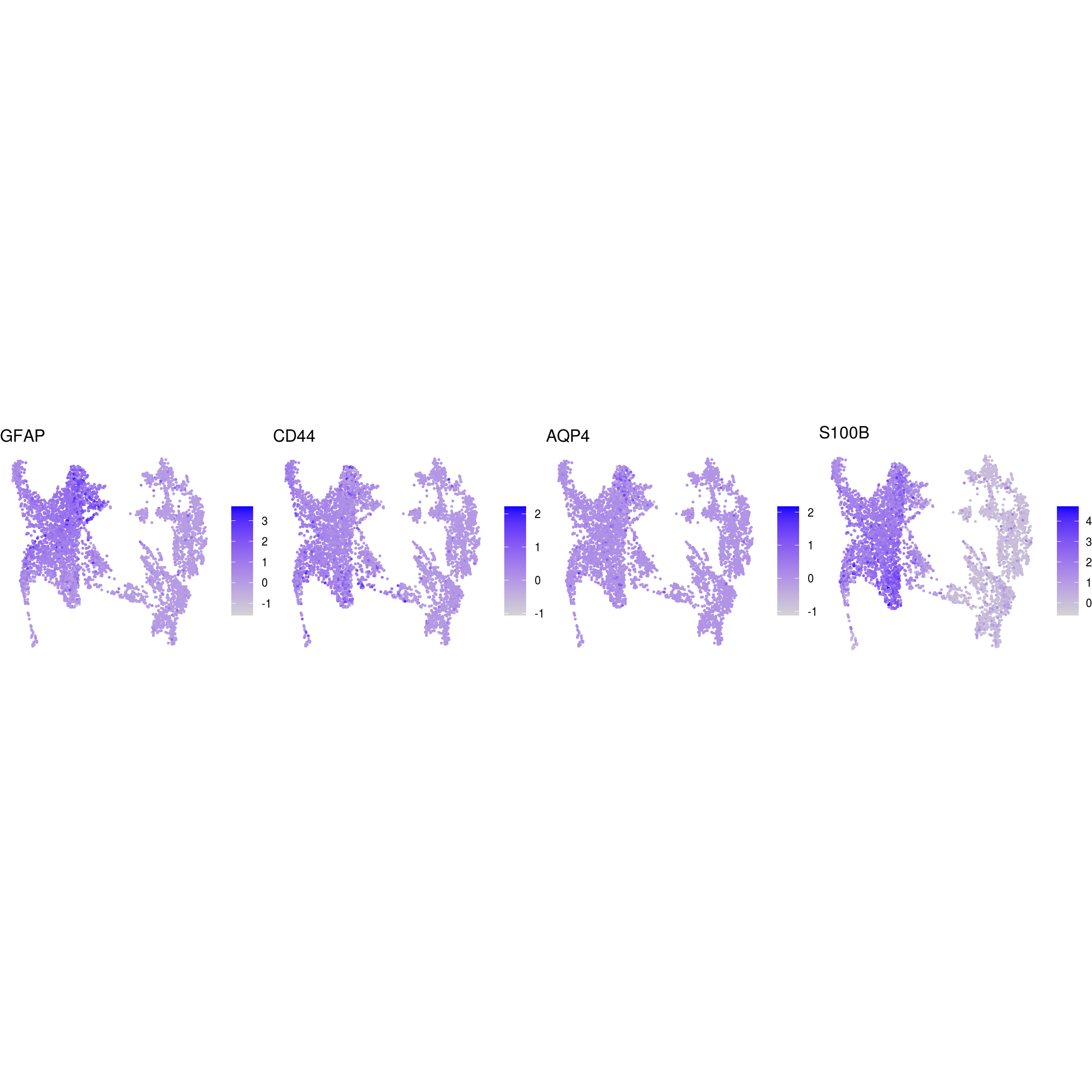
DR colored by marker expression
# downsample to 5000 cells
cs <- sample(colnames(sce), 5e3)
sub <- subset(so, cells = cs)
# UMAPs colored by marker-expression
for (m in seq_along(fs)) {
cat("## ", names(fs)[m], "\n")
ps <- lapply(seq_along(fs[[m]]), function(i) {
if (!fs[[m]][i] %in% rownames(so)) return(NULL)
FeaturePlot(sub, features = fs[[m]][i], reduction = "umap", pt.size = 0.4) +
theme(aspect.ratio = 1, legend.position = "none") +
ggtitle(labs[[m]][i]) + theme_void() + theme(aspect.ratio = 1)
})
# arrange plots in grid
ps <- ps[!vapply(ps, is.null, logical(1))]
p <- plot_grid(plotlist = ps, ncol = 4, label_size = 10)
print(p)
cat("\n\n")
}
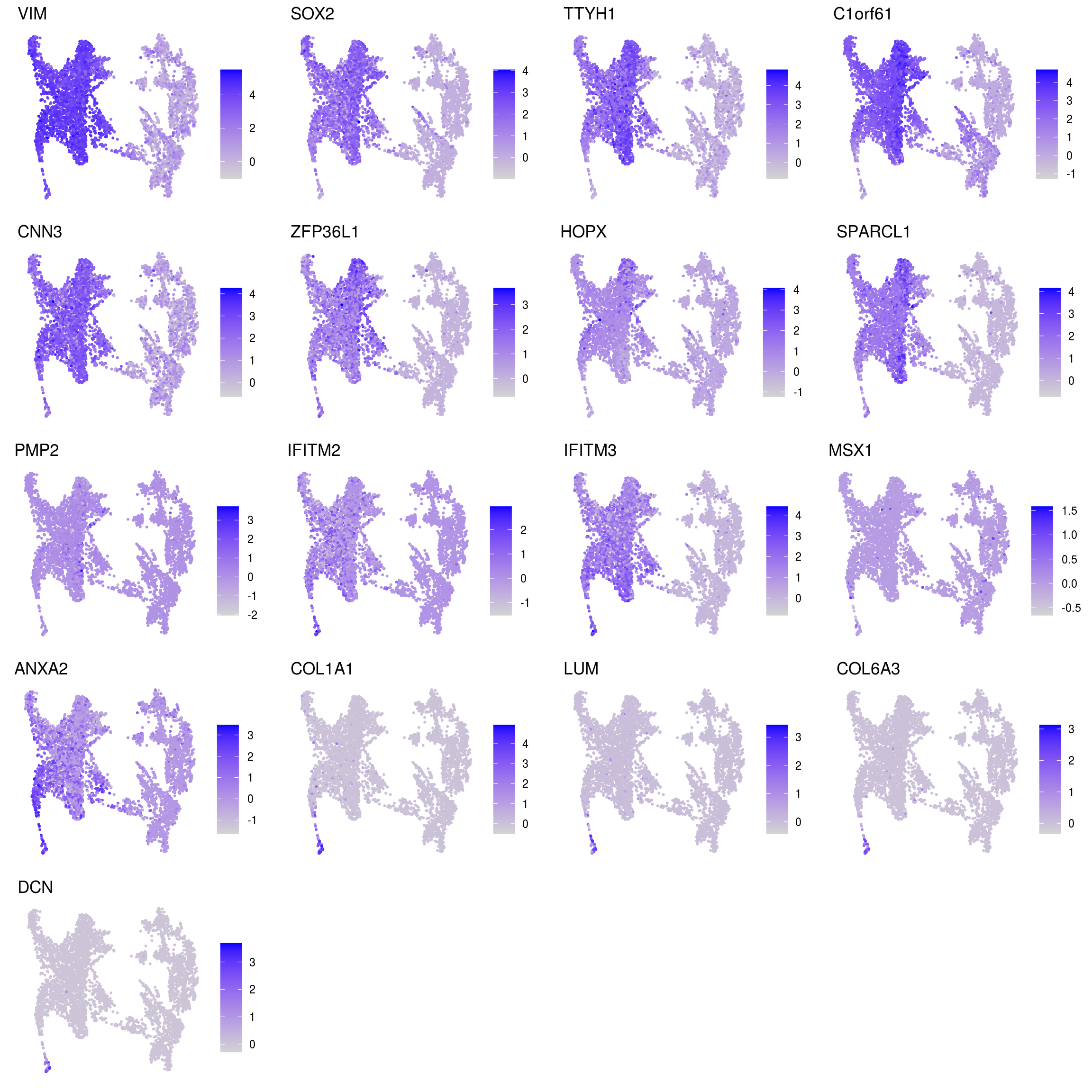
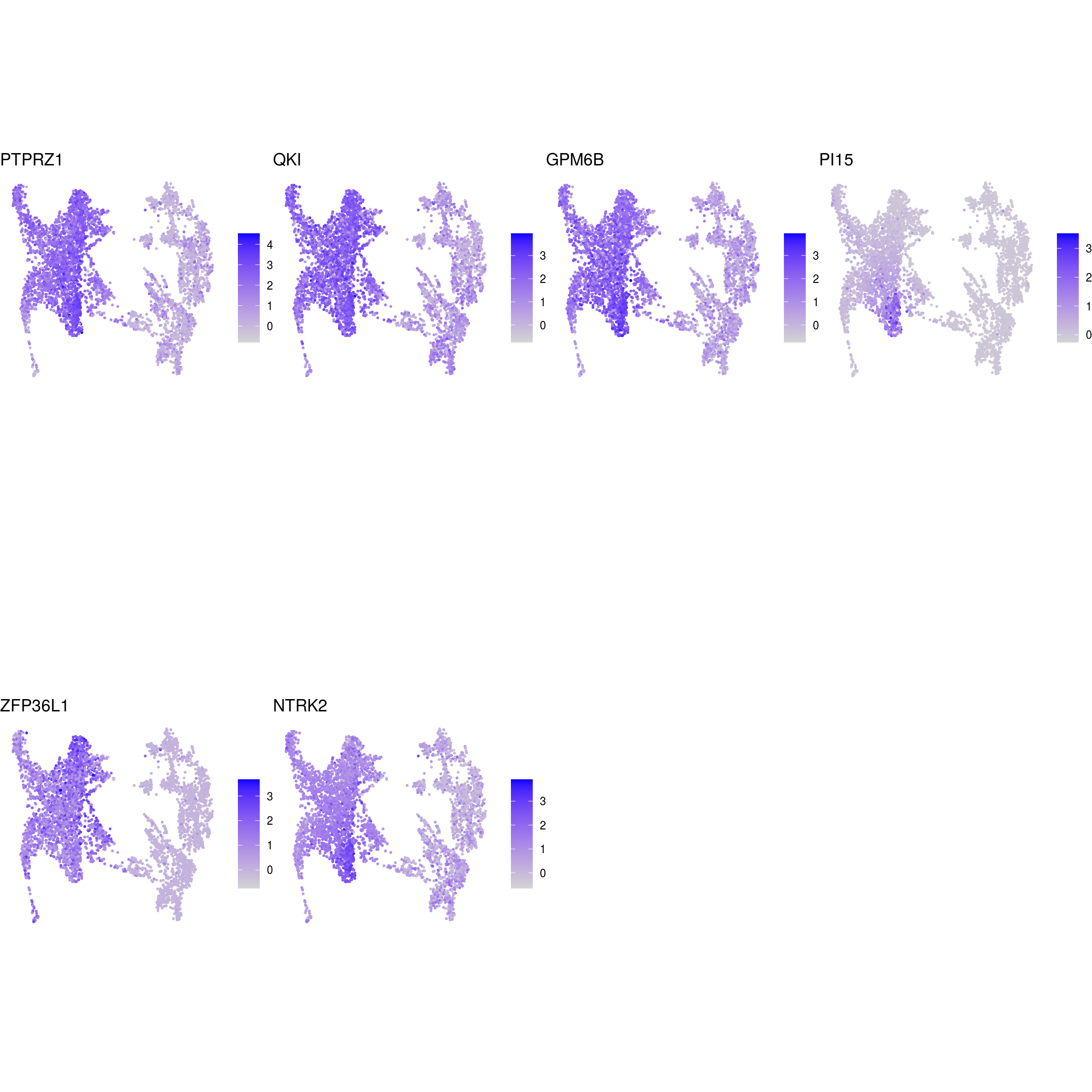
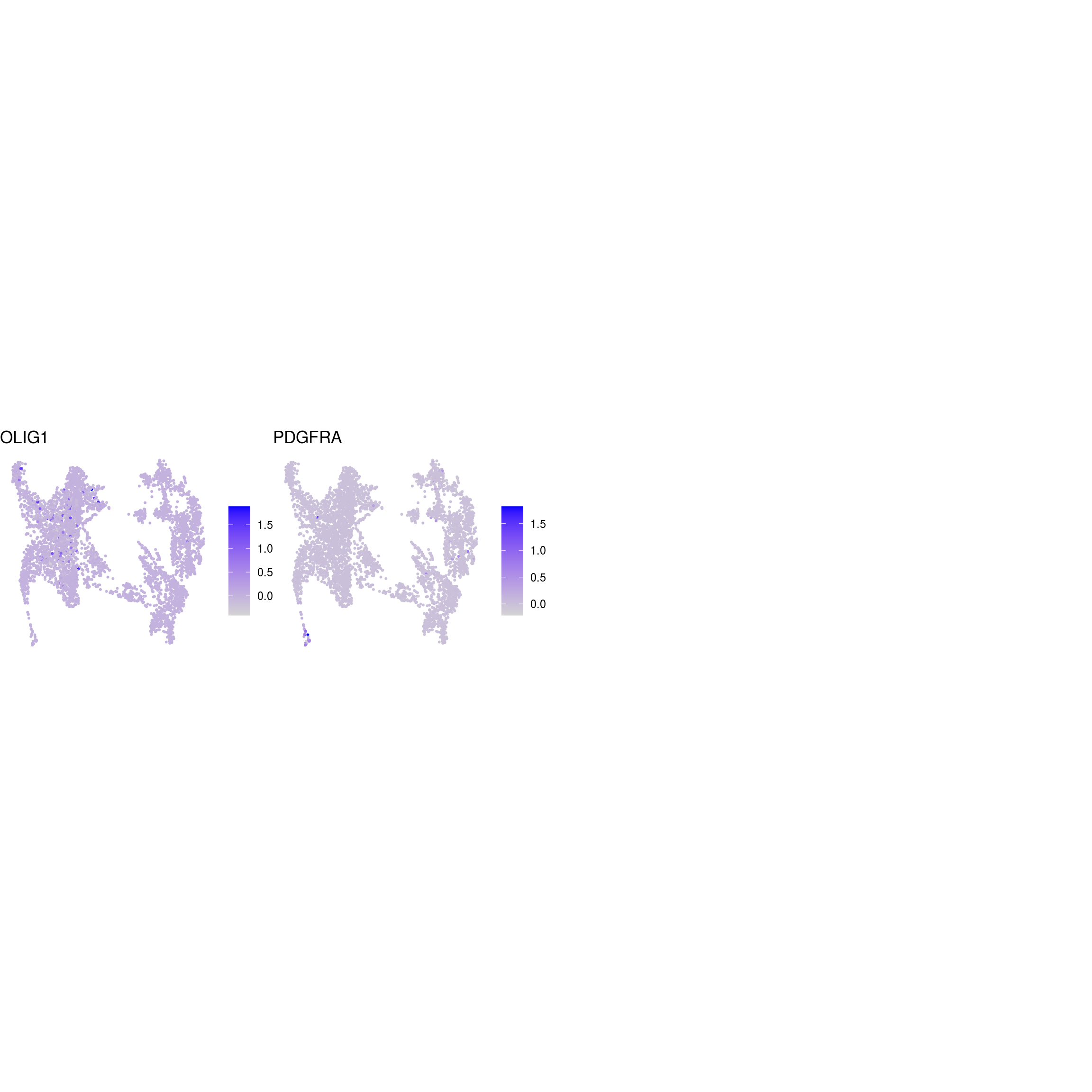
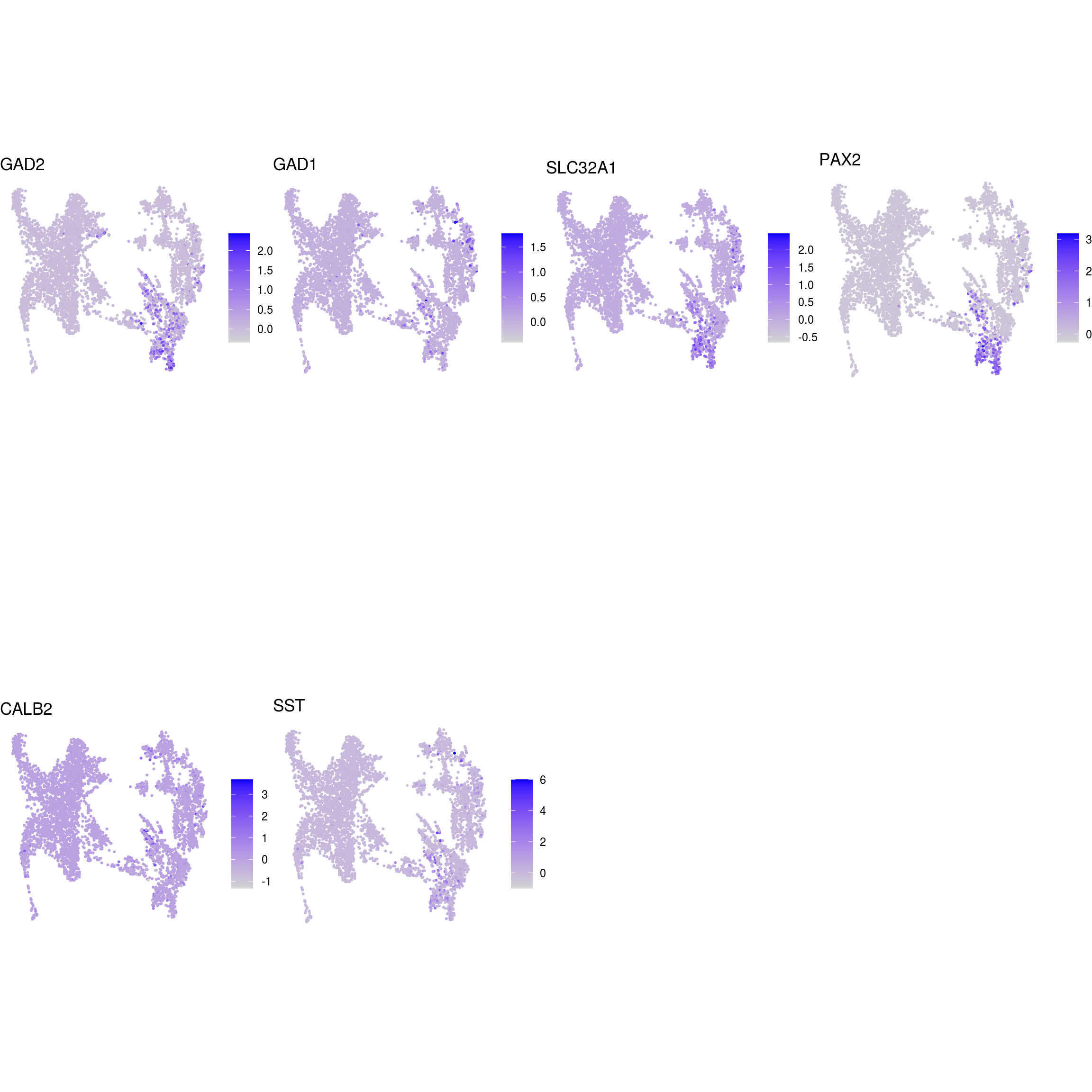
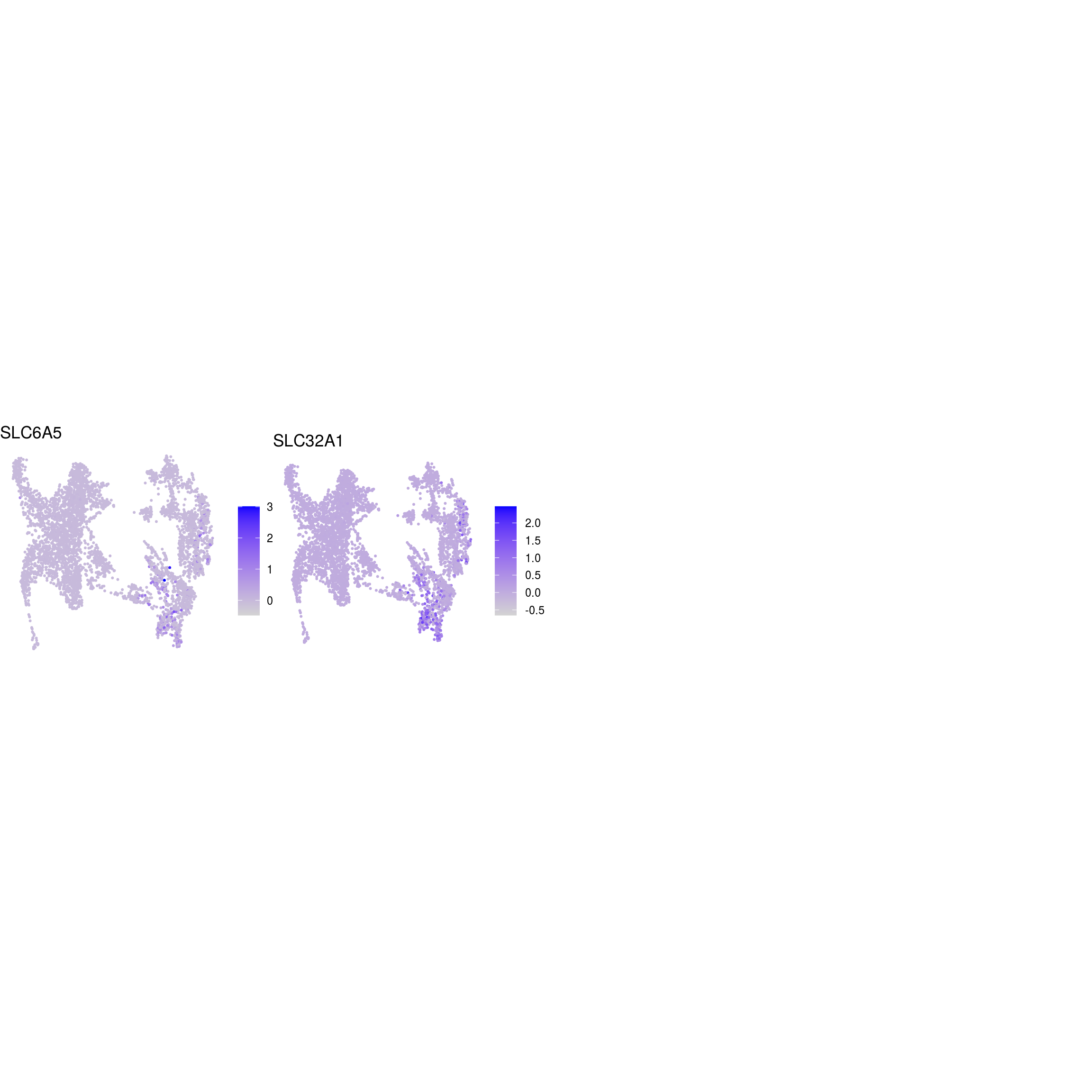

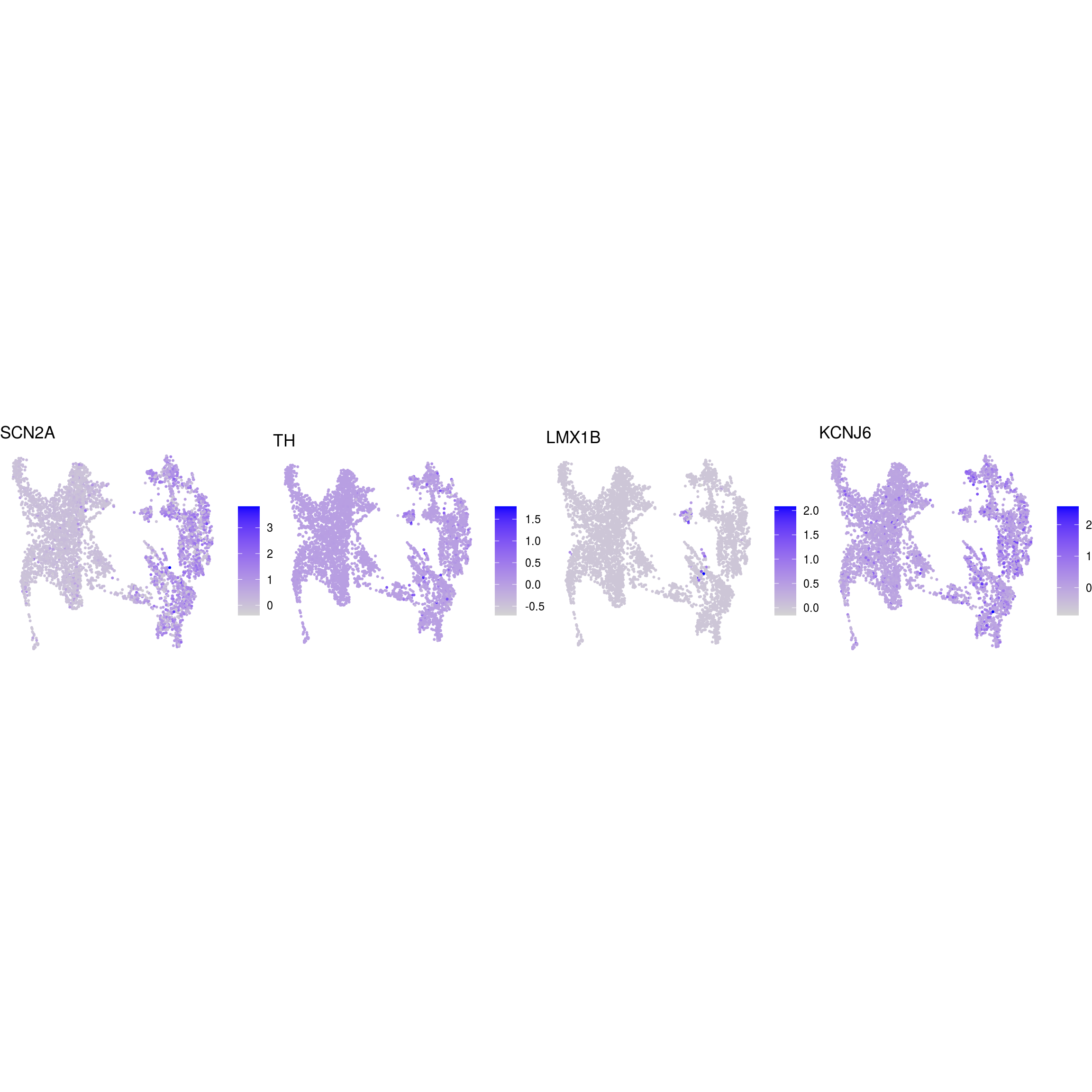
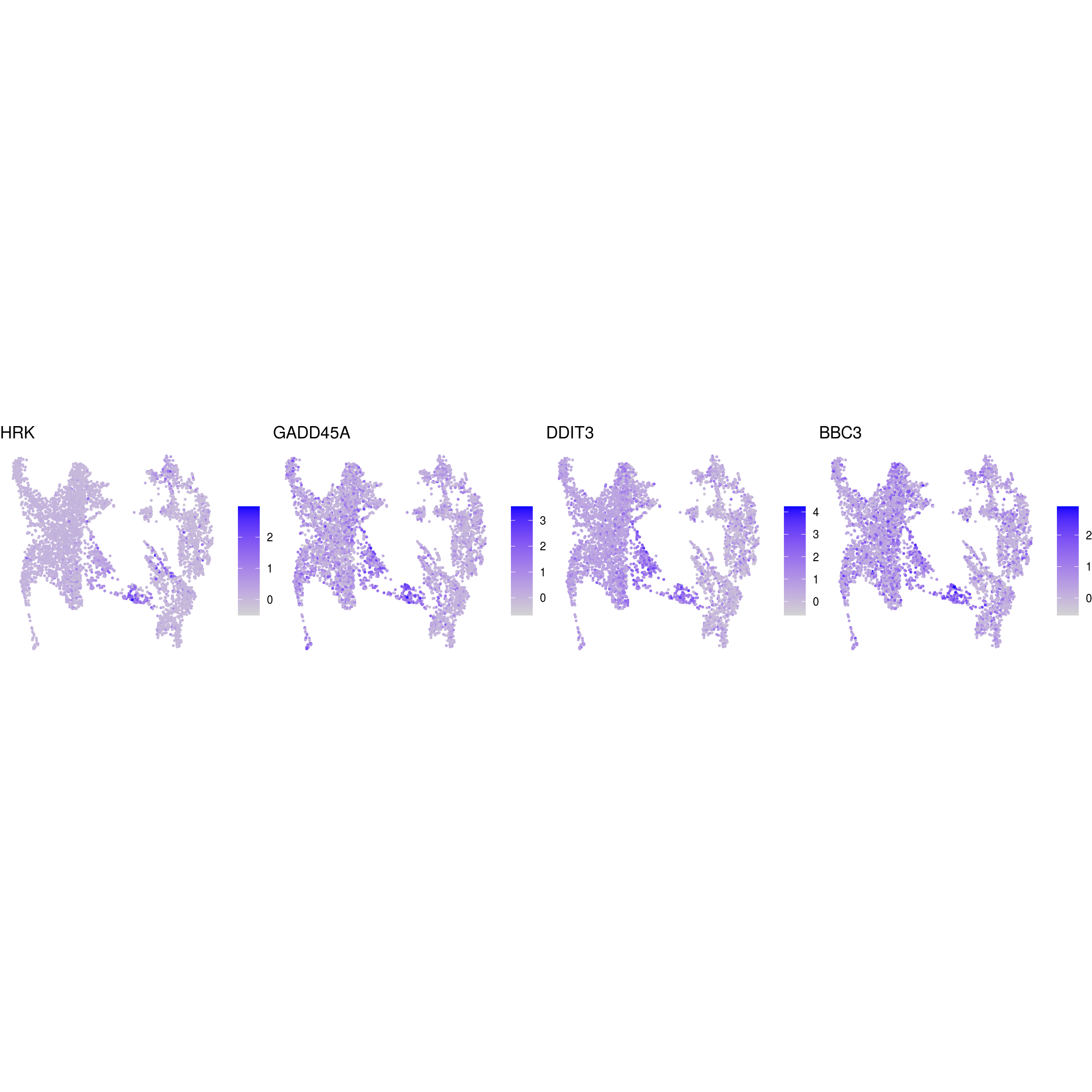
Cluster annotation
Based on the plots we annotated the clusters: …
sessionInfo()R version 4.0.0 (2020-04-24)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 16.04.6 LTS
Matrix products: default
BLAS: /usr/local/R/R-4.0.0/lib/libRblas.so
LAPACK: /usr/local/R/R-4.0.0/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 grid stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] BiocParallel_1.22.0 RCurl_1.98-1.2
[3] stringr_1.4.0 Seurat_3.1.5
[5] scran_1.16.0 SingleCellExperiment_1.10.1
[7] SummarizedExperiment_1.18.1 DelayedArray_0.14.0
[9] matrixStats_0.56.0 Biobase_2.48.0
[11] GenomicRanges_1.40.0 GenomeInfoDb_1.24.0
[13] IRanges_2.22.2 S4Vectors_0.26.1
[15] BiocGenerics_0.34.0 viridis_0.5.1
[17] viridisLite_0.3.0 RColorBrewer_1.1-2
[19] purrr_0.3.4 muscat_1.2.0
[21] dplyr_0.8.5 ggplot2_3.3.0
[23] cowplot_1.0.0 ComplexHeatmap_2.4.2
[25] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] backports_1.1.7 circlize_0.4.9
[3] blme_1.0-4 igraph_1.2.5
[5] plyr_1.8.6 lazyeval_0.2.2
[7] TMB_1.7.16 splines_4.0.0
[9] listenv_0.8.0 scater_1.16.0
[11] digest_0.6.25 foreach_1.5.0
[13] htmltools_0.4.0 gdata_2.18.0
[15] lmerTest_3.1-2 magrittr_1.5
[17] memoise_1.1.0 cluster_2.1.0
[19] doParallel_1.0.15 ROCR_1.0-11
[21] limma_3.44.1 globals_0.12.5
[23] annotate_1.66.0 prettyunits_1.1.1
[25] colorspace_1.4-1 rappdirs_0.3.1
[27] ggrepel_0.8.2 blob_1.2.1
[29] xfun_0.14 jsonlite_1.6.1
[31] crayon_1.3.4 genefilter_1.70.0
[33] lme4_1.1-23 zoo_1.8-8
[35] ape_5.3 survival_3.1-12
[37] iterators_1.0.12 glue_1.4.1
[39] gtable_0.3.0 zlibbioc_1.34.0
[41] XVector_0.28.0 leiden_0.3.3
[43] GetoptLong_0.1.8 BiocSingular_1.4.0
[45] future.apply_1.5.0 shape_1.4.4
[47] scales_1.1.1 DBI_1.1.0
[49] edgeR_3.30.0 Rcpp_1.0.4.6
[51] xtable_1.8-4 progress_1.2.2
[53] clue_0.3-57 reticulate_1.16
[55] dqrng_0.2.1 bit_1.1-15.2
[57] rsvd_1.0.3 tsne_0.1-3
[59] htmlwidgets_1.5.1 httr_1.4.1
[61] gplots_3.0.3 ellipsis_0.3.1
[63] ica_1.0-2 farver_2.0.3
[65] pkgconfig_2.0.3 XML_3.99-0.3
[67] uwot_0.1.8 locfit_1.5-9.4
[69] labeling_0.3 tidyselect_1.1.0
[71] rlang_0.4.6 reshape2_1.4.4
[73] later_1.0.0 AnnotationDbi_1.50.0
[75] munsell_0.5.0 tools_4.0.0
[77] RSQLite_2.2.0 ggridges_0.5.2
[79] evaluate_0.14 yaml_2.2.1
[81] knitr_1.28 bit64_0.9-7
[83] fs_1.4.1 fitdistrplus_1.1-1
[85] caTools_1.18.0 RANN_2.6.1
[87] pbapply_1.4-2 future_1.17.0
[89] nlme_3.1-148 whisker_0.4
[91] pbkrtest_0.4-8.6 compiler_4.0.0
[93] plotly_4.9.2.1 beeswarm_0.2.3
[95] png_0.1-7 variancePartition_1.18.0
[97] tibble_3.0.1 statmod_1.4.34
[99] geneplotter_1.66.0 stringi_1.4.6
[101] lattice_0.20-41 Matrix_1.2-18
[103] nloptr_1.2.2.1 vctrs_0.3.0
[105] pillar_1.4.4 lifecycle_0.2.0
[107] lmtest_0.9-37 GlobalOptions_0.1.1
[109] RcppAnnoy_0.0.16 BiocNeighbors_1.6.0
[111] data.table_1.12.8 bitops_1.0-6
[113] irlba_2.3.3 patchwork_1.0.0
[115] httpuv_1.5.2 colorRamps_2.3
[117] R6_2.4.1 promises_1.1.0
[119] KernSmooth_2.23-17 gridExtra_2.3
[121] vipor_0.4.5 codetools_0.2-16
[123] boot_1.3-25 MASS_7.3-51.6
[125] gtools_3.8.2 assertthat_0.2.1
[127] DESeq2_1.28.1 rprojroot_1.3-2
[129] rjson_0.2.20 withr_2.2.0
[131] sctransform_0.2.1 GenomeInfoDbData_1.2.3
[133] hms_0.5.3 tidyr_1.1.0
[135] glmmTMB_1.0.1 minqa_1.2.4
[137] rmarkdown_2.1 DelayedMatrixStats_1.10.0
[139] Rtsne_0.15 git2r_0.27.1
[141] numDeriv_2016.8-1.1 ggbeeswarm_0.6.0