Cell cycle scoring
Katharina Hembach
6/18/2020
Last updated: 2020-06-26
Checks: 7 0
Knit directory: neural_scRNAseq/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200522) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version afe57cc. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: ._.DS_Store
Ignored: ._MA.pdf
Ignored: ._MA2.pdf
Ignored: ._MA_plots.pdf
Ignored: ._Rplots.pdf
Ignored: .__workflowr.yml
Ignored: ._hm.pdf
Ignored: ._neural_scRNAseq.Rproj
Ignored: ._sample5_MA_2nd_pop.pdf
Ignored: ._sample5_QC_2nd_pop.pdf
Ignored: ._tmp.pdf
Ignored: ._tmp_detected.pdf
Ignored: ._tmp_manual_discard.pdf
Ignored: ._tmp_manual_discard1.pdf
Ignored: ._tmp_manual_discard_all.pdf
Ignored: ._tmp_manual_discard_all1.pdf
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/._.DS_Store
Ignored: analysis/._01-preprocessing.Rmd
Ignored: analysis/._01-preprocessing.html
Ignored: analysis/._02.1-SampleQC.Rmd
Ignored: analysis/._03-filtering.Rmd
Ignored: analysis/._04-clustering.Rmd
Ignored: analysis/._04-clustering.knit.md
Ignored: analysis/._04.1-cell_cycle.Rmd
Ignored: analysis/._05-annotation.Rmd
Ignored: analysis/.__site.yml
Ignored: analysis/._additional_filtering.Rmd
Ignored: analysis/._additional_filtering_clustering.Rmd
Ignored: analysis/._index.Rmd
Ignored: analysis/01-preprocessing_cache/
Ignored: analysis/02-1-SampleQC_cache/
Ignored: analysis/02-quality_control_cache/
Ignored: analysis/02.1-SampleQC_cache/
Ignored: analysis/03-filtering_cache/
Ignored: analysis/04-clustering_cache/
Ignored: analysis/05-annotation_cache/
Ignored: analysis/additional_filtering_cache/
Ignored: analysis/additional_filtering_clustering_cache/
Ignored: analysis/sample5_QC_cache/
Ignored: data/.DS_Store
Ignored: data/._.DS_Store
Ignored: data/._.smbdeleteAAA17ed8b4b
Ignored: data/._metadata.csv
Ignored: data/data_sushi/
Ignored: data/filtered_feature_matrices/
Ignored: output/.DS_Store
Ignored: output/._.DS_Store
Ignored: output/additional_filtering.rds
Ignored: output/figures/
Ignored: output/sce_01_preprocessing.rds
Ignored: output/sce_02_quality_control.rds
Ignored: output/sce_03_filtering.rds
Ignored: output/sce_preprocessing.rds
Ignored: output/so_04_1_cell_cycle.rds
Ignored: output/so_04_clustering.rds
Ignored: output/so_additional_filtering_clustering.rds
Untracked files:
Untracked: MA.pdf
Untracked: MA2.pdf
Untracked: MA_plots.pdf
Untracked: Rplots.pdf
Untracked: analysis/additional_filtering.Rmd
Untracked: analysis/additional_filtering_clustering.Rmd
Untracked: analysis/sample5_QC.Rmd
Untracked: analysis/tabsets.Rmd
Untracked: hm.pdf
Untracked: sample5_MA_2nd_pop.pdf
Untracked: sample5_QC_2nd_pop.pdf
Untracked: scripts/
Untracked: tmp.pdf
Untracked: tmp_detected.pdf
Untracked: tmp_manual_discard.pdf
Untracked: tmp_manual_discard1.pdf
Untracked: tmp_manual_discard_all.pdf
Untracked: tmp_manual_discard_all1.pdf
Unstaged changes:
Modified: analysis/_site.yml
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/04.1-cell_cycle.Rmd) and HTML (docs/04.1-cell_cycle.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 0983178 | khembach | 2020-06-26 | Fix tabset |
| html | 06330b1 | khembach | 2020-06-22 | Build site. |
| Rmd | f349423 | khembach | 2020-06-21 | regress out number of UMIs and perc mitochondrial features; cyclone |
Load packages
library(cowplot)
library(ggplot2)
library(RColorBrewer)
library(viridis)
library(scran)
library(Seurat)
library(SingleCellExperiment)
library(stringr)
library(RCurl)
library(BiocParallel)
library(dplyr)Load data & convert to SCE
so <- readRDS(file.path("output", "so_04_clustering.rds"))
sce <- as.SingleCellExperiment(so, assay = "RNA")
colData(sce) <- as.data.frame(colData(sce)) %>%
mutate_if(is.character, as.factor) %>%
DataFrame(row.names = colnames(sce))
so <- SetIdent(so, value = "integrated_snn_res.0.4")
so@meta.data$cluster_id <- Idents(so)
sce$cluster_id <- Idents(so)Cell cycle scoring with Seurat
We assign each cell a cell cycle scores and visualize them in the DR plots. We use known G2/M and S phase markers that come with the Seurat package. The markers are anticorrelated and cells that to not express the markers should be in G1 phase.
We compute cell cycle phase:
DefaultAssay(so) <- "RNA"
# A list of cell cycle markers, from Tirosh et al, 2015
cc_file <- getURL("https://raw.githubusercontent.com/hbc/tinyatlas/master/cell_cycle/Homo_sapiens.csv")
cc_genes <- read.csv(text = cc_file)
# match the marker genes to the features
m <- match(cc_genes$geneID[cc_genes$phase == "S"],
str_split(rownames(GetAssayData(so)),
pattern = "\\.", simplify = TRUE)[,1])
s_genes <- rownames(GetAssayData(so))[m]
(s_genes <- s_genes[!is.na(s_genes)]) [1] "ENSG00000012963.UBR7" "ENSG00000049541.RFC2"
[3] "ENSG00000051180.RAD51" "ENSG00000073111.MCM2"
[5] "ENSG00000075131.TIPIN" "ENSG00000076003.MCM6"
[7] "ENSG00000076248.UNG" "ENSG00000077514.POLD3"
[9] "ENSG00000092470.WDR76" "ENSG00000092853.CLSPN"
[11] "ENSG00000093009.CDC45" "ENSG00000094804.CDC6"
[13] "ENSG00000095002.MSH2" "ENSG00000100297.MCM5"
[15] "ENSG00000101868.POLA1" "ENSG00000104738.MCM4"
[17] "ENSG00000111247.RAD51AP1" "ENSG00000112312.GMNN"
[19] "ENSG00000117748.RPA2" "ENSG00000118412.CASP8AP2"
[21] "ENSG00000119969.HELLS" "ENSG00000129173.E2F8"
[23] "ENSG00000131153.GINS2" "ENSG00000132646.PCNA"
[25] "ENSG00000132780.NASP" "ENSG00000136492.BRIP1"
[27] "ENSG00000136982.DSCC1" "ENSG00000143476.DTL"
[29] "ENSG00000144354.CDCA7" "ENSG00000151725.CENPU"
[31] "ENSG00000156802.ATAD2" "ENSG00000159259.CHAF1B"
[33] "ENSG00000162607.USP1" "ENSG00000163950.SLBP"
[35] "ENSG00000167325.RRM1" "ENSG00000168496.FEN1"
[37] "ENSG00000171848.RRM2" "ENSG00000174371.EXO1"
[39] "ENSG00000175305.CCNE2" "ENSG00000176890.TYMS"
[41] "ENSG00000197299.BLM" "ENSG00000198056.PRIM1"
[43] "ENSG00000276043.UHRF1" m <- match(cc_genes$geneID[cc_genes$phase == "G2/M"],
str_split(rownames(GetAssayData(so)),
pattern = "\\.", simplify = TRUE)[,1])
g2m_genes <- rownames(GetAssayData(so))[m]
(g2m_genes <- g2m_genes[!is.na(g2m_genes)]) [1] "ENSG00000010292.NCAPD2" "ENSG00000011426.ANLN"
[3] "ENSG00000013810.TACC3" "ENSG00000072571.HMMR"
[5] "ENSG00000075218.GTSE1" "ENSG00000080986.NDC80"
[7] "ENSG00000087586.AURKA" "ENSG00000088325.TPX2"
[9] "ENSG00000089685.BIRC5" "ENSG00000092140.G2E3"
[11] "ENSG00000094916.CBX5" "ENSG00000100401.RANGAP1"
[13] "ENSG00000102974.CTCF" "ENSG00000111665.CDCA3"
[15] "ENSG00000112742.TTK" "ENSG00000113810.SMC4"
[17] "ENSG00000114346.ECT2" "ENSG00000115163.CENPA"
[19] "ENSG00000117399.CDC20" "ENSG00000117650.NEK2"
[21] "ENSG00000117724.CENPF" "ENSG00000120802.TMPO"
[23] "ENSG00000123485.HJURP" "ENSG00000123975.CKS2"
[25] "ENSG00000126787.DLGAP5" "ENSG00000129195.PIMREG"
[27] "ENSG00000131747.TOP2A" "ENSG00000134222.PSRC1"
[29] "ENSG00000134690.CDCA8" "ENSG00000136108.CKAP2"
[31] "ENSG00000137804.NUSAP1" "ENSG00000137807.KIF23"
[33] "ENSG00000138160.KIF11" "ENSG00000138182.KIF20B"
[35] "ENSG00000138778.CENPE" "ENSG00000139354.GAS2L3"
[37] "ENSG00000142945.KIF2C" "ENSG00000143228.NUF2"
[39] "ENSG00000143401.ANP32E" "ENSG00000143815.LBR"
[41] "ENSG00000148773.MKI67" "ENSG00000157456.CCNB2"
[43] "ENSG00000158402.CDC25C" "ENSG00000164104.HMGB2"
[45] "ENSG00000169607.CKAP2L" "ENSG00000169679.BUB1"
[47] "ENSG00000170312.CDK1" "ENSG00000173207.CKS1B"
[49] "ENSG00000175063.UBE2C" "ENSG00000175216.CKAP5"
[51] "ENSG00000178999.AURKB" "ENSG00000184661.CDCA2"
[53] "ENSG00000188229.TUBB4B" "ENSG00000189159.JPT1" so <- CellCycleScoring(so, s.features = s_genes, g2m.features = g2m_genes,
set.ident = TRUE)
DefaultAssay(so) <- "integrated"Cell cycle assignment using cyclone
## read pretrained set of human cell cycle markers
human_pairs <- readRDS(system.file("exdata", "human_cycle_markers.rds",
package="scran"))
# Using Ensembl IDs to match up with the annotation in 'mm.pairs'.
assignments <- cyclone(sce, human_pairs,
gene.names = str_split(rownames(sce), pattern = "\\.",
simplify = TRUE)[,1],
BPPARAM = MulticoreParam(workers = 20),
verbose = TRUE)
table(assignments$phases, colData(sce)$cluster_id)
0 1 2 3 4 5 6 7 8 9 10 11 12 13
G1 7717 1745 678 2019 2415 1247 1232 1214 1684 1039 957 950 747 472
G2M 637 241 1581 136 26 149 89 92 30 99 101 37 5 33
S 2840 1952 597 608 167 1085 1146 1026 234 732 282 341 424 471
14 15 16
G1 601 401 197
G2M 15 14 27
S 299 73 93## Add cell cycle phases to Seurat object
so$cyclone_phase <- assignments$phasesColored DR
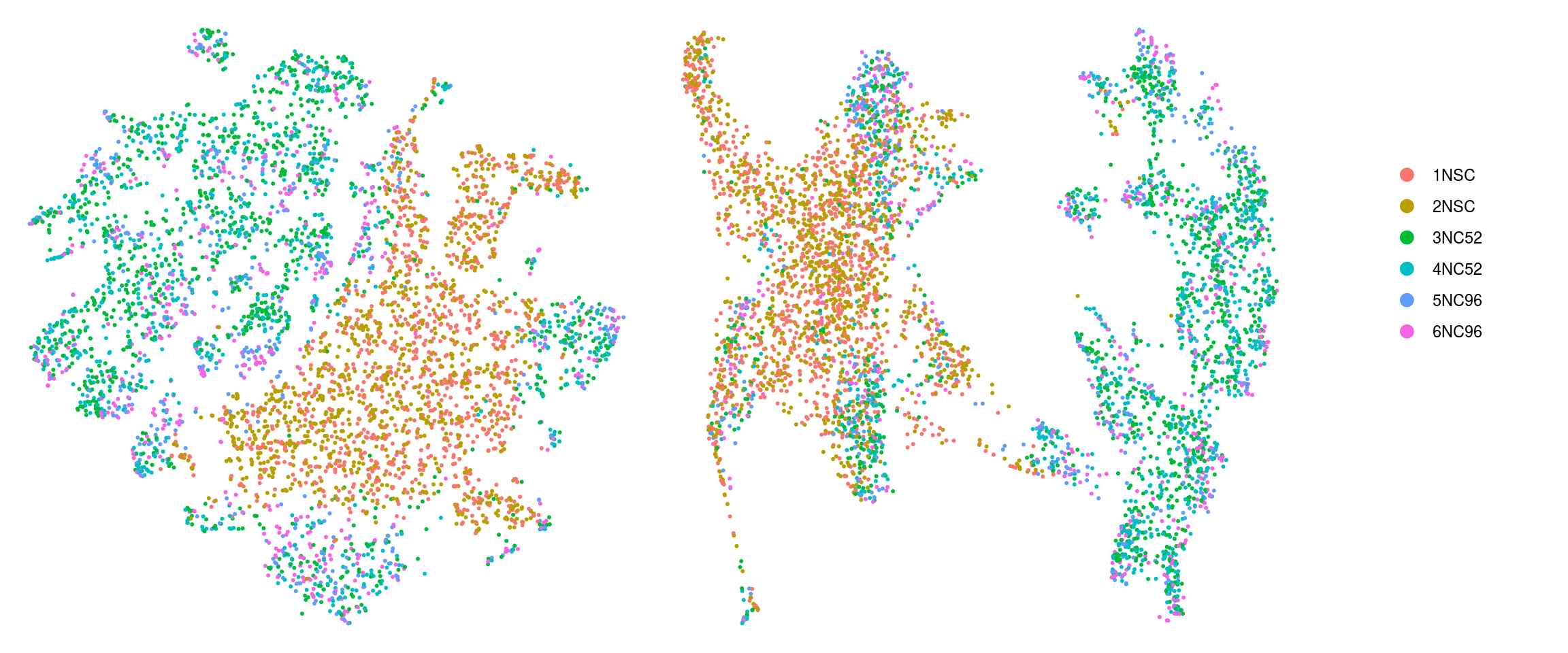
cs <- sample(colnames(so), 5e3)
.plot_dr <- function(so, dr, id)
DimPlot(so, cells = cs, group.by = id, reduction = dr, pt.size = 0.4) +
guides(col = guide_legend(nrow = 11,
override.aes = list(size = 3, alpha = 1))) +
theme_void() + theme(aspect.ratio = 1)
ids <- c("cluster_id", "group_id", "sample_id", "Phase", "cyclone_phase")
for (id in ids) {
cat("## ", id, "\n")
p1 <- .plot_dr(so, "tsne", id)
lgd <- get_legend(p1)
p1 <- p1 + theme(legend.position = "none")
p2 <- .plot_dr(so, "umap", id) + theme(legend.position = "none")
ps <- plot_grid(plotlist = list(p1, p2), nrow = 1)
p <- plot_grid(ps, lgd, nrow = 1, rel_widths = c(1, 0.2))
print(p)
cat("\n\n")
}



Save Seurat object to RDS
saveRDS(so, file.path("output", "so_04_1_cell_cycle.rds"))
sessionInfo()R version 4.0.0 (2020-04-24)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 16.04.6 LTS
Matrix products: default
BLAS: /usr/local/R/R-4.0.0/lib/libRblas.so
LAPACK: /usr/local/R/R-4.0.0/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] dplyr_0.8.5 BiocParallel_1.22.0
[3] RCurl_1.98-1.2 stringr_1.4.0
[5] Seurat_3.1.5 scran_1.16.0
[7] SingleCellExperiment_1.10.1 SummarizedExperiment_1.18.1
[9] DelayedArray_0.14.0 matrixStats_0.56.0
[11] Biobase_2.48.0 GenomicRanges_1.40.0
[13] GenomeInfoDb_1.24.0 IRanges_2.22.2
[15] S4Vectors_0.26.1 BiocGenerics_0.34.0
[17] viridis_0.5.1 viridisLite_0.3.0
[19] RColorBrewer_1.1-2 ggplot2_3.3.0
[21] cowplot_1.0.0 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] Rtsne_0.15 ggbeeswarm_0.6.0
[3] colorspace_1.4-1 ellipsis_0.3.1
[5] ggridges_0.5.2 rprojroot_1.3-2
[7] XVector_0.28.0 BiocNeighbors_1.6.0
[9] fs_1.4.1 farver_2.0.3
[11] leiden_0.3.3 listenv_0.8.0
[13] ggrepel_0.8.2 codetools_0.2-16
[15] splines_4.0.0 knitr_1.28
[17] scater_1.16.0 jsonlite_1.6.1
[19] ica_1.0-2 cluster_2.1.0
[21] png_0.1-7 uwot_0.1.8
[23] sctransform_0.2.1 compiler_4.0.0
[25] httr_1.4.1 dqrng_0.2.1
[27] backports_1.1.7 lazyeval_0.2.2
[29] assertthat_0.2.1 Matrix_1.2-18
[31] limma_3.44.1 later_1.0.0
[33] BiocSingular_1.4.0 htmltools_0.4.0
[35] tools_4.0.0 rsvd_1.0.3
[37] igraph_1.2.5 gtable_0.3.0
[39] glue_1.4.1 GenomeInfoDbData_1.2.3
[41] reshape2_1.4.4 RANN_2.6.1
[43] rappdirs_0.3.1 Rcpp_1.0.4.6
[45] vctrs_0.3.0 ape_5.3
[47] nlme_3.1-148 DelayedMatrixStats_1.10.0
[49] lmtest_0.9-37 xfun_0.14
[51] globals_0.12.5 lifecycle_0.2.0
[53] irlba_2.3.3 statmod_1.4.34
[55] future_1.17.0 edgeR_3.30.0
[57] zlibbioc_1.34.0 MASS_7.3-51.6
[59] zoo_1.8-8 scales_1.1.1
[61] promises_1.1.0 yaml_2.2.1
[63] reticulate_1.16 pbapply_1.4-2
[65] gridExtra_2.3 stringi_1.4.6
[67] rlang_0.4.6 pkgconfig_2.0.3
[69] bitops_1.0-6 evaluate_0.14
[71] lattice_0.20-41 ROCR_1.0-11
[73] purrr_0.3.4 labeling_0.3
[75] htmlwidgets_1.5.1 patchwork_1.0.0
[77] tidyselect_1.1.0 RcppAnnoy_0.0.16
[79] plyr_1.8.6 magrittr_1.5
[81] R6_2.4.1 pillar_1.4.4
[83] whisker_0.4 withr_2.2.0
[85] fitdistrplus_1.1-1 survival_3.1-12
[87] tsne_0.1-3 tibble_3.0.1
[89] future.apply_1.5.0 crayon_1.3.4
[91] KernSmooth_2.23-17 plotly_4.9.2.1
[93] rmarkdown_2.1 locfit_1.5-9.4
[95] grid_4.0.0 data.table_1.12.8
[97] git2r_0.27.1 digest_0.6.25
[99] tidyr_1.1.0 httpuv_1.5.2
[101] munsell_0.5.0 beeswarm_0.2.3
[103] vipor_0.4.5